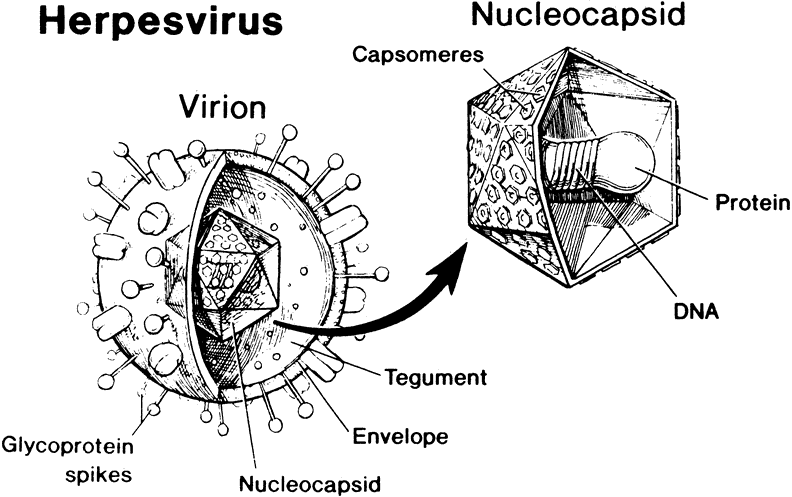
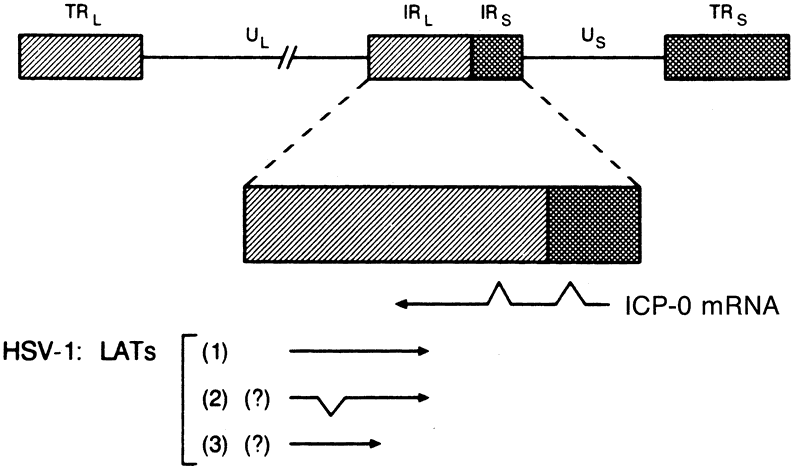
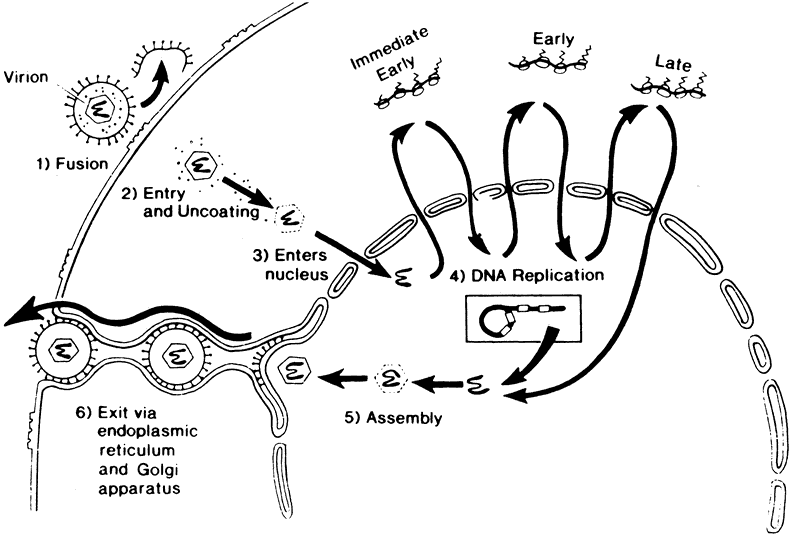
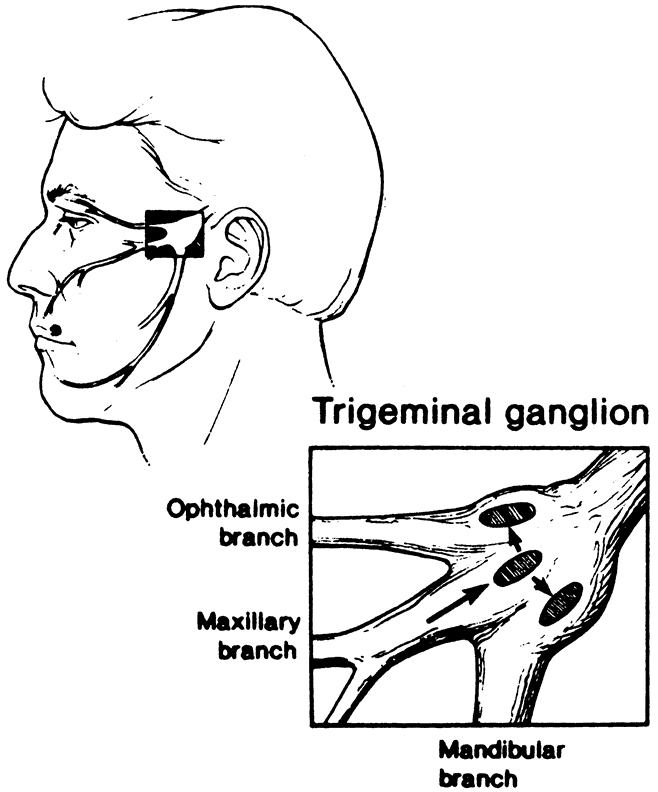
1. Liesegang TJ: Herpes simplex virus epidemiology and ocular importance. Cornea 20:1, 2001 2. Liesegang TJ, Melton LJ, Daly PJ et al: Epidemiology of ocular herpes simplex. Incidence in Rochester, MN, 1950 through 1982. Arch Ophthalmol 107:1155, 1989 3. Margolis TP, Atherton SS: Herpes simplex virus diseases: Posterior segment
of the eye. In Pepose JS, Holland GN, Wilhelmus KR (eds): Ocular Infection
and Immunity. St Louis: Mosby, 1996:1155 4. Roizman B: Herpesviridae. In Fields BN, Knipe DM, Howley PM, et al (eds): Fields
Virology. Philadelphia: Lippincott-Raven, 1996:2221 5. Liesegang TJ: Biology and molecular aspects of herpes simplex and varicella-zoster virus
infections. Ophthalmology 99:781, 1992 6. Roizman B, Sears AE: Herpes simplex viruses and their replication. In Fields
BN, Knipe DM, Howley PM, et al (eds): Fields Virology. Philadelphia: Lippincott-Raven, 1996:2231 7. Spear PG: Glycoproteins specified by herpes simplex virus. In Roizman B (ed): The
Herpesviruses. New York: Plenum, 1985:315 8. McGeoch DJ, Dolan A, Donald S et al: Sequence determination and genetic content of the short unique region in
the genome of herpes simplex virus type 1. J Mol Biol 181:1, 1985 9. McGeoch DJ, Dalrymple MA, Davision AF et al: The complete DNA sequence of the long unique region in the genome of herpes
simplex virus type 1. J Gen Virol 69:1531, 1988 10. Buchman TG, Simpson T, Nosal C et al: The structure of herpes simplex virus DNA and its application to molecular
epidemiology. Ann N Y Acad Sci 354:279, 1980 11. Centifanto-Fitzgerald YM, Yamaguchi T, Kaufman HE et al: Ocular disease pattern induced by herpes simplex virus is genetically determined
by a specific region of viral DNA. J Exp Med 155:475, 1982 12. Smeraglia R, Hochadel J, Varnell ED et al: The role of herpes simplex virus secreted glycoproteins in herpetic keratitis. Exp Eye Res 35:443, 1982 13. Tullo AB, Coupes D, Klapper P et al: Analysis of glycoproteins expressed by isolates of herpes simplex virus
causing different forms of keratitis in man. Curr Eye Res 6:33, 1987 14. Dix RD, McKendall RR, Baringer JR: Comparative neurovirulence of herpes simplex virus type 1 strains after
peripheral or intracerebral inoculation of BALB/c mice. Infect Immun 40:103, 1983 15. Thompson RL, Cook ML, Devi-Rao GB et al: Functional and molecular analysis of the avirulent wild-type herpes simplex
virus type 1 strain KOS. J Virol 58:203, 1986 16. Thompson RL, Devi-Rao GV, Stevens JG et al: Rescue of a herpes simplex virus type 1 neurovirulence function with a
cloned DNA fragment. J Virol 55:504, 1985 17. Thompson RL, Rogers SK, Zerhusen MA: Herpes simplex virus neurovirulence and productive infection of neural
cells is associated with a function which maps between 0.82 and 0.832 map
units on the HSV genome. Virology 172:435, 1989 18. Chou J, Kern E, Whitley RJ et al: Mapping of neurovirulence to protein 31.5 encoded by a diploid herpes simplex
virus type 1 gene nonessential for growth in cell culture. Science 250:1262, 1990 19. Whitley RJ, Kern ER, Chatterjee S et al: Replication, establishment of latency, and induced reactivation of herpes
simplex virus γ1 34.5 deletion mutants in rodent models. J Clin Invest 91:2837, 1993 20. Chou J, Chen JJ, Gross M et al: Association of Mr 90,000 phosphoprotein with protein kinase PKR in cells exhibiting enhanced
phosphorylation of translation initiation factor eIF-2α and premature
shutoff of protein synthesis after infection with γ134.5- mutants of herpes simplex virus 1. Proc Natl Acad Sci U S A 92:10516, 1995 21. Black FL: Infectious diseases in primitive societies. Science 187:515, 1975 22. Whitley RJ: Herpes simplex viruses. In Fields BN, Knipe DM, Howley PM, et
al (eds): Fields Virology. Philadelphia: Lippincott-Raven, 1996:2297 23. Klein RJ: Pathogenetic mechanisms of recurrent herpes simplex virus infections. Arch Virol 51:1, 1976 24. Fenwick M, Morse LS, Roizman B: Anatomy of herpes simplex virus DNA. XI. Apparent clustering of functions
effecting rapid inhibition of host DNA and protein synthesis. J Virol 29:825, 1979 25. Honess RW, Roizman B: Regulation of herpevirus macromolecular synthesis. Part I. Cascade regulation
of synthesis of viral proteins. J Virol 14:8, 1974 26. McKnight JLC, Kristie TM, Roizman B: Binding of the virion protein mediating α gene induction in herpes
simplex virus 1-infected cells to its cis site requires cellular proteins. Proc Natl Acad Sci U S A 84:7061, 1987 27. Jacob RJ, Roizman B: Anatomy of herpes simplex virus DNA. XIII. Accumulation of head to tail
concatamers in nuclei of infected cells and their role in the generation
of the four isomeric arrangement of viral DNA. J Virol 23:394, 1977 28. Katz JP, Boden ET, Coen DM: Quantitative polymerase chain reaction analysis of herpes simplex virus
DNA in ganglia of mice infected with replication incompetent mutants. J Virol 64:4288, 1990 29. Wagner EK, Bloom DC: Experimental investigation of herpes simplex virus latency. Clin Microbiol Rev 10:419, 1997 30. Wagner EK, Bloom DC: HSV gene expression during latent infection and reactivation. In
Medveczky PG, Friedman H, Bendinelli M (eds): Herpesviruses
and Immunity. New York: Plenum, 1998:53 31. Ellerick DM, Fraser NW: Physical state of the latent herpes simplex virus genome in a mouse model
system: Evidence suggesting an episomal state. Virology 158:265, 1987 32. Dix RD, Mills J: Experimental mouse models of herpes simplex virus infection. In
Zak O, Sande MA (eds): Experimental Models in Antimicrobial
Chemotherapy. Vol 2. London: Academic, 1986:219 33. Lillycrop KA, Dent CL, Wheatley S et al: The octamer-binding protein Oct-2 represses HSV immediate-early genes in
cell lines derived from latently infectible sensory neuron. Neuron 7:381, 1991 34. Stevens JG, Wagner EK, Devi-Rao GB et al: RNA complementary to a herpesvirus α gene mRNA is prominent in latently
infected neurons. Science 235:1056, 1987 35. Stevens JG, Haarr L, Porter DD et al: Prominence of the herpes simplex virus latency-associated transcript in
trigeminal ganglia from seropositive humans. J Infect Dis 158:117, 1988 36. Krause PR, Croen KD, Straus SE et al: Detection and preliminary characterization of herpes simplex virus type 1 transcripts
in latently infected human trigeminal ganglia. J Virol 62:4819, 1988 37. Fraser NW, Block TM, Spivack JG: The latency-associated transcripts of herpes simplex virus: RNA in search
of function. Virology 191:1, 1992 38. Farrel MJ, Dobson AT, Feldmen LT: Herpes simplex virus latency-associated transcript is a stable intron. Proc Natl Acad Sci U S A 88:790, 1991 39. Rock DL, Nesburn AB, Ghiasi H et al: Detection of latency related viral RNAs in trigeminal ganglia of rabbits
latently infected with herpes simplex virus type 1. J Virol 61:3820, 1987 40. Perng G-C, Jones C, Ciacci-Zanella J et al: Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated
transcript. Science 287:1500, 2000 41. Inman M, Perng G-C, Herderson G et al: Region of herpes simplex virus type 1 latency-associated transcript sufficient
for wild-type spontaneous reactivation promotes cell survival
in tissue culture. J Virol 75:3636, 2001 42. Thompson RL, Sawtell NM: Herpes simplex virus type 1 latency-associated transcript gene promotes
neuronal survival. J Virol 75:6660, 2001 43. Ahmed M, Lock M, Miller CG et al: Regions of the herpes simplex virus type 1 latency-associated transcript
that protect cells from apoptosis in vitro and protect neuronal cells
in vivo. J Virol 76:717, 2002 44. Henderson S, Huen D, Rowe M et al: Epstein-Barr virus-coded BHRF1 protein, a viral homologue of Bcl-2, protects
human B-cells from programmed cell death. Proc Natl Acad Sci U S A 90:8479, 1993 45. Derfuss T, Fickenscher H, Kraft MS et al: Antiapoptotic activity of the herpesvirus saimiri-encoded Bcl-2 homolog: Stabilization
of mitochondria and inhibition of caspase 3-like activity. J Virol 72:5897, 1998 46. Aubert M, Blaho J: The herpes simplex virus type 1 regulatory protein ICP27 is required for
the prevention of apoptosis in infected human cells. J Virol 73:2803, 1999 47. Ho DY, Mocarski ES: Herpes simplex virus latent RNA (LAT) is not required for latent infection
in the mouse. Proc Natl Acad Sci U S A 86:7596, 1989 48. Sederati F, Izumi KM, Wagner EK et al: Herpes simplex virus type 1 latency associated transcription plays no role
in establishment or maintenance of a latent infection in murine sensory
neurons. J Virol 63:4455, 1989 49. Halford WP, Schaffer PA: ICP0 is required for efficient reactivation of herpes simplex virus type 1 from
neuronal latency. J Virol 75:3240, 2001 50. Abghari SZ, Stulting RD: Recovery of herpes simplex virus from ocular tissues of latently infected
inbred mice. Invest Ophthalmol Vis Sci 29:239, 1988 51. Kaye SM, Lynas C, Patterson A et al: Evidence for herpes simplex viral latency in the human cornea. Br J Ophthalmol 75:195, 1991 52. Easty DL: Herpes simplex virus isolation in chronic stromal keratitis, human and
laboratory studies. Curr Eye Res 6:69, 1987 53. Laylock KA, Lee SF, Stulting RD et al: Herpes simplex virus type 1 transcription is not detectable in quiescent
human stromal keratitis by in situ hybridization. Invest Ophthalmol Vis Sci 34:285, 1993 54. Cleator GM, Klapper PE, Dennett C et al: Corneal donor infection by herpes simplex virus: Herpes simplex virus DNA
in donor corneas. Cornea 13:294, 1994 55. Neufeld MV, Steinemann TL, Merin LM et al: Identification of a herpes simplex virus-induced dendrite in an eye-bank
donor cornea. Cornea 18:489, 1999 56. Redpath S, Angulo A, Gascoigne NRJ: Immune checkpoints in viral latency. Annu Rev Microbiol 55:531, 2001 57. McKendall RR, Klassen T, Baringer JR: Host defenses in herpes simplex infections of the nervous system: Effect
of antibody on disease and viral spread. Infect Immun 23:305, 1979 58. Oakes JE, Lausch RN: Role of Fc fragments in antibody-mediated recovery from ocular and subcutaneous
herpes simplex virus infections. Infect Immun 33:109, 1981 59. Dix RD, Pereira L, Baringer JR: Use of monoclonal antibody directed against herpes simplex virus glycoproteins
to protect mice against acute virus-induced neurological disease. Infect Immun 34:192, 1981 60. Rector ZT, Lausch RN, Oakes JE: Use of monoclonal antibodies for analysis of antibody-dependent immunity
to ocular herpes simplex virus type 1 infection. Infect Immun 38:168, 1982 61. Rector JR, Lausch RN, Oakes JE: Identification of infected cell-specific monoclonal antibodies and their
role in host resistance to ocular herpes simplex virus type 1 infection. J Gen Virol 65:657, 1984 62. Dix RD: Glycoprotein gB of herpes simplex virus expresses type-common and type-specific
antigenic determinants in vivo. J Med Virol 30:192, 1990 63. Dix RD, Mills J: Acute and latent herpes simplex virus neurological disease in mice immunized
with purified virus-specific glycoproteins gB or gD. J Med Virol 17:9, 1985 64. Blyth WA, Harbour DA, Hill TJ: Pathogenesis of zosterform spread of herpes simplex virus in the mouse. J Gen Virol 65:1477, 1984 65. Schmid DS, Rouse BT: The role of T cell immunity in control of herpes simplex virus. Curr Top Microbiol Immunol 179:57, 1992. 66. Smith G: Virus strategies for evasion of the host response to infection. Trends Microbiol 2:81, 1994 67. York IA, Roop C, Andrews DW et al: A cytosolic herpes simplex virus protein inhibits antigen presentation
to CD8+ T lymphocytes. Cell 77:525, 1994 68. Hill A, Jugovic P, York I et al: Herpes simplex virus turns off the TAP to evade host immunity. Nature 375:411, 1995 69. Jones TR, Hanson LK, Sun L et al: Multiple independent loci within the human cytomegalovirus unique short
region down-regulate expression of major histocompatibility complex class
I heavy chains. J Virol 69:4830, 1995 70. Moore K, Vieira P, Fiorentino D et al: Homology of cytokine synthesis inhibitory factor (Il-10) to the Epstein-Barr
virus gene BCRF1. Science 248:1230, 1990 71. Sundmacher R: Clinical aspects of herpetic eye diseases. Curr Eye Res 6:183, 1987 72. Pepose JS, Leib DA, Stuart M et al: Herpes simplex virus diseases: Anterior
segment of the eye. In Pepose JS, Holland GN, Wilhelmus KR (eds): Ocular
Infection and Immunity. St Louis: Mosby, 1996:905 73. Hendricks RL: Immunopathogenesis of viral ocular infections. Chem Immunol 73:120, 1999 74. Thomas J. Rouse BT: Immunopathogenesis of herpetic ocular disease. Immunol Res 16:375, 1997 75. Stulting RD, Kindle JC, Nahmias AJ: Patterns of herpes simplex keratitis in inbred mice. Invest Ophthalmol Vis Sci 26:1360, 1985 76. Chen W, Tang Q, Hendricks RL: Ex vivo model of leukocytes migrate into herpes simplex virus-infected
mouse corneas. J Leukoc Biol 60:167, 1996 77. Thomas J, Gangappa S, Kanangat S et al: On the essential involvement of neutrophils in the immunopathologic disease: Herpetic
stromal keratitis. J Immunol 158:1383, 1997 78. Tumpey TM, Chen SH, Oakes JE et al: Neutrophil-mediated suppression of virus replication after herpes simplex
virus type 1 infection of the murine cornea. J Virol 70:898, 1996 79. Hendricks RL, Tumpey TM, Finnegan A: IFN-gamma and IL-2 are protective in the skin but pathologic in the corneas
of HSV-1-infected mice. J Immunol 149:3023, 1992 80. Zheng M, Deshpande S, Lee S et al: Contribution of vascular endothelial growth factor in the neovascularization
process during the pathogenesis of herpetic stromal keratitis. J Virol 75:9828, 2001 81. Metcalf JF, Hamilton DS, Reichert RW: Herpetic keratitis in athymic (nude) mice. Infect Immun 26:1164, 1979 82. Brandt CR: Susceptibility of +/+, +/v, and v/v BALB/c mice to ocular herpes simplex virus infection. Ophthalmic Res 24:332, 1992 83. Mercadal C, Bouley D, DeStephano D et al: Herpetic stromal keratitis in the reconstituted SCID mouse model. J Virol 67:3404, 1993 84. Russel RG, Naisse MP, Larsen HS et al: Role of T lymphocytes in the pathogenesis of herpetic stromal keratitis. Invest Ophthalmol Vis Sci 25:938, 1984 85. Von Herranth MG, Oldstone MBA: Virus-induced autoimmune disease. Curr Opin Immunol 8:878, 1996 86. Oldstone MBA: Molecular mimicry and immune-mediated diseases. FASEB J 12:1255, 1998 87. Whitton LJ, Fujinami RS: Viruses as triggers of autoimmunity: Facts and fantasies. Curr Opin Microbiol 2:392, 1999 88. Sercarz EE, Lehmann PV, Ametani A et al: Dominance and crypticity of T cell antigenic determinants. Annu Rev Immunol 11:729, 1993 89. Zhou A, Granucci F, Yeh L et al: Molecular mimicry by herpes simplex virus type-1: Autoimmune disease after
viral infection. Science 279:1344, 1998 90. Avery AC, Zhao ZS, Rodriguez A et al: Resistance to herpes stromal keratitis conferred by an IgG2a derived peptide. Nature 276:431, 1995 91. Opremcak EM, Wells PA, Thompson P et al: Immunogenetic influence of IgH-1 phenotype on experimental herpes simplex
virus type-1 corneal infection. Invest Ophthalmol Vis Sci 29:749, 1988 92. Thomas J, Rouse BT: Immunopathology of herpetic stromal keratitis: Discordance in CD4+ T
cell function between euthymic host and reconstituted SCID recipients. J Immunol 160:3965, 1998 93. Deshpande SP, Lee S, Zheng M et al: Herpes simplex virus-induced keratitis: Evaluation of the role of molecular
mimicry in lesion pathogenesis. J Virol 75:3077, 2001 94. Ghiasi H, Slanina S, Nesburn AB et al: Characterization of baculovirus-expressed herpes simplex virus type 1 glycoprotein
K. J Virol 68:2347, 1994 95. Ghiasi H, Kaiwar R, Nesburn AB et al: Immunoselection of recombinant baculoviruses expressing high levels of
biologically active herpes simplex virus type 1 glycoprotein D. Arch Virol 121:163, 1991 96. Ghiasi H, Kaiwar R, Nesburn AB et al: Expression of glycoprotein B of herpes simplex virus type 1 in insect cells: Analysis
of its biochemical and immunological properties. Virus Res 22:25, 1992 97. Ghiasi H, Kaiwar R, Nesburn AB et al: Baculovirus expressed glycoprotein G of herpes simplex virus type 1 partially
protects vaccinated mice against lethal HSV-1 challenge. Virology 190:233, 1992 98. Ghiasi H, Kaiwar R, Nesburn AB et al: Expression of herpes simplex virus type 1 glycoprotein I (gI) in baculovirus: Preliminary
biochemical characterization and protective studies. J Virol 66:2505, 1992 99. Ghiasi H, Kaiwar R, Nesburn AB et al: Baculovirus expressed glycoprotein H (gH) of herpes simplex virus type 1 (HSV-1) induces
neutralizing antibody and delayed hypersensitivity responses
but does not protect immunized mice against lethal HSV-1 challenge. J Gen Virol 73:719, 1992 100. Ghiasi H, Kaiwar R, Nesburn AB et al: Baculovirus expressed herpes simplex virus type 1 glycoprotein C protects
mice from lethal HSV-1 infection. Antiviral Res 18:291, 1992 101. Ghiasi H, Kaiwar R, Nesburn AB et al: Baculovirus expressed glycoprotein E (gE) of herpes simplex virus type-1 (HSV-1) protects
mice against lethal intraperitoneal and lethal ocular
HSV-1 challenge. Virology 188:469, 1992 102. Ghiasi H, Cai S, Nesburn AB et al: Vaccination with herpes simplex virus type 1 glycoprotein K impairs clearance
of virus from the trigeminal ganglia resulting in chronic infection. Virology 224:330, 1996 103. Ghiasi H, Cai S, Slanina S et al: Nonneutralizing antibody against the glycoprotein K of herpes simplex virus
type-1 exacerbates herpes simplex virus type-1-induced corneal scarring
in various virus-mouse strain combinations. Invest Ophthalmol Vis Sci 38:1213, 1997 104. Ghiasi H, Hofman FM, Cai S et al: Vaccination with different HSV-1 glycoproteins induces different patterns
of ocular cytokine responses following HSV-1 challenge of vaccinated
mice. Vaccine 17:2576, 1999 105. Ghiasi H, Perng G-C, Nesburn AB et al: Antibody-dependent enhancement of HSV-1 infection by anti-gK sera. Virus Res 68:137, 2000 106. Littaua R, Kurane I, Ennis FA: Human IgG Fc receptor II mediates antibody-mediated enhancement of dengue
virus infection. J Immunol 144:3183, 1990 107. Halstead SB: In vivo enhancement of dengue virus infection in rhesus monkeys by passively
transferred antibody. J Infect Dis 140:527, 1979 108. Culbertson WW, Dix RD: Varicella-zoster virus diseases: Posterior segment
of the eye. In Pepose JS, Holland GN, Wilhelmus KR (eds): Ocular Infection
and Immunity. St Louis: Mosby, 1996:1131 109. Lewis ML, Culbertson WW, Post JD et al: Herpes simplex virus type 1. A cause of acute retinal necrosis syndrome. Ophthalmology 96:875, 1989 110. Duker JS, Nielsen JC, Eagle RC Jr et al: Rapidly progressive acute retinal necrosis secondary to herpes simplex
virus, type 1. Ophthalmology 97:1638, 1990 111. De La Paz MA, Young LHY: Acute retinal necrosis syndrome. Semin Ophthalmol 8:61, 1993 112. el Azazi, Samuelsson A, Linde A et al: Intrathecal antibody production against viruses of the herpesvirus family
in acute retinal necrosis syndrome. Am J Ophthalmol 112:76, 1991 113. von Szily A: Experimental endogenous transmission of infection from bulbus to bulbus. Klin Monatsbl Augenheilkd 75:593, 1924 114. Whittum JA, McCulley JP, Niederkorn JY et al: Ocular disease induced in mice by anterior chamber inoculation of herpes
simplex virus. Invest Ophthalmol Vis Sci 25:1065, 1984 115. Hamasaki DI, Dix RD, Atherton SS: Bilateral alterations of the ERG and retinal histology following unilateral
HSV-1 inoculation. Invest Ophthalmol Vis Sci 29:1242, 1988 116. Atherton SS, Streilein SW: Two waves of virus following anterior chamber inoculation HSV-1. Invest Ophthalmol Vis Sci 28:571, 1987 117. Margolis TP, LaVail JH, Setzer PY et al: Selective spread of herpes simplex virus in the central nervous system
after ocular infection. J Virol 63:4756, 1989 118. Vann VR, Atherton SS: Neural spread of herpes simplex virus after anterior chamber inoculation. Invest Ophthalmol Vis Sci 32:2462, 1991 119. Olson RM, Holland GN, Goss SJ et al: Routes of viral spread in the von Szily model of herpes simplex virus retinopathy. Curr Eye Res 6:59, 1987 120. Cousins SW, Gonzalez A, Atherton SS: Herpes simplex retinitis in the mouse: Clinicopathologic correlations. Invest Ophthalmol Vis Sci 30:1485, 1989 121. Azumi A, Cousins SW, Kanter MY et al: Modulation of murine herpes simplex virus type 1 retinitis in the uninoculated
eye by CD4+ T lymphocytes. Invest Ophthalmol Vis Sci 35:54, 1994 122. Azumi A, Atherton SS: Sparing of the ipsilateral retina following anterior chamber inoculation
of HSV-1: Requirement for either CD4+ or CD8+ T cells. Invest Ophthalmol Vis Sci 35:3251, 1994 123. Berra A, Rodriguez A, Heiligenhaus A et al: The role of macrophages in the pathogenesis of HSV-1 induced chorioretinitis
in BALB/c mice. Invest Ophthalmol Vis Sci 35:2990, 1994 124. Streilein JW, Igietseme JU, Atherton SS: Evidence that precursor cytotoxic T cells mediate acute necrosis in HSV-1-infected
retinas. Curr Eye Res 10:81, 1991 125. Zaltas MM, Opremcak M, Hemady R et al: Immunohistopathologic findings in herpes simplex virus chorioretinitis
in the von Szily model. Invest Ophthalmol Vis Sci 33:68, 1992 126. Atherton SS, Altman NH, Streilein JW: Histopathologic study of herpes virus-induced retinitis in athymic BALB/c
mice: Evidence for an immunopathologic process. Curr Eye Res 8:1179, 1989 127. Cousins SW, Altman NH, Atherton SS: Schisis contributes to necrosis in experimental HSV-1 retinitis. Exp Eye Res 48:745, 1987 128. Igietseme JU, Calzada PJ, Gonzalez AR et al: Protection of mice from herpes simplex virus-induced retinitis by in vitro-activated
immune cells. J Virol 63:4808, 1989 129. Zhao M, Azumi A, Atherton SS: T lymphocyte infiltration in the brain following anterior chamber inoculation
of HSV-1. J Neuroimmunol 58:11, 1995 130. Atherton SS: Protection from retinal necrosis by passive transfer of monoclonal antibody
specific for herpes simplex virus glycoprotein D. Curr Eye Res 11:45, 1992 131. Zhao M, Atherton SS: Immune effector cell (IEC)-mediated protection from HSV-1 retinitis occurs
in the brain. J Neuroimmunol 75:51, 1997 132. Dix RD, Hamasaki DI, Hurst L et al: Bilateral retinal disease following unilateral inoculation of mice with
a HSV-1 isolate from a patient with acute retinal necrosis. Invest Ophthalmol Vis Sci 31:314, 1990 133. Streilein JW: Regional immunity of the eye. In Pepose JS, Holland GN, Wilhelmus
KR (eds): Ocular Infection and Immunity. St Louis: Mosby, 1996:19 134. Niederkorn JY, Ferguson TA: Anterior chamber associated immune deviation (ACAID). In
Pepose JS, Holland GN, Wilhelmus KR (eds): Ocular Infection
and Immunity. St Louis: Mosby, 1996:96 135. Streilein JW, Atherton SS, Vann V: A critical role for ACAID in the distinctive pattern of retinitis that
follows anterior chamber inoculation of HSV-1. Curr Eye Res 6:127, 1987 136. Kielty D, Cousins SW, Atherton SS: HSV-1 retinitis and delayed hypersensitivity in DBA/2 and C57BL/6 mice. Invest Ophthalmol Vis Sci 28:1994, 1987 137. Chodosh J, Banks MC, Stroop WG: Rose bengal inhibits herpes simplex virus replication in Vero and human
corneal epithelial cells in vitro. Invest Ophthalmol Vis Sci 33:2520, 1992 138. Brooks SE, Kaza V, Nakamura T et al: Photo inactivation of herpes simplex virus by rose bengal and fluorescein. In
vitro and in vivo studies. Cornea 13:43, 1994 139. Chodosh J, Dix RD, Howell RC et al: Staining characteristics and antiviral activity of sulforhodamine B and
lissamine green B. Invest Ophthalmol Vis Sci 35:1046, 1994 140. Stroop WG, Chen MC, Chodosh J et al: Polymerase chain reaction assessment of herpes simplex virus type 1 corneal
infection in animals treated with rose bengal and lissamine green
B. Invest Ophthalmol Vis Sci 41:2096, 2000 141. Waldrep JC, Mondino BJ: Humoral immunity and the eye. In Pepose JS, Holland
GN, Wilhelmus KR (eds): Ocular Infection and Immunity. St Louis: Mosby, 1996:33 142. Goldmann H, Witmer R: Antikurper in Kammerwasser. Ophthalmologica 177:323, 1954 143. Witmer R: Clinical implications of aqueous humor studies in uveitis. Am J Ophthalmol 86:39, 1978 144. Mullis K, Faloona F, Scharf S et al: Specific enzymatic amplification of DNA in vitro: The polymerase chain
reaction. Symp Quant Biol 51:263, 1986 145. Fox GM, Crouse CA, Chuang EL et al: Detection of herpevirus DNA in vitreous and aqueous specimens by the polymerase
chain reaction. Arch Ophthalmol 109:266, 1991 146. Grant DM: Acyclovir (Zovirax) ophthalmic ointment: A review of clinical tolerance. Curr Eye Res 6:231, 1987 147. Charles SJ, Gray JJ: Ocular herpes simplex virus infection: Reduced sensitivity to acyclovir
in primary disease. Br J Ophthalmol 74:286, 1990 148. Sonkin PL, Baratz KH, Frothingham R et al: Acyclovir-resistant herpes simplex virus keratouveitis after penetrating
keratoplasty. Ophthalmology 99:1805, 1992 149. Dix RD: Prospects for a vaccine against herpes simplex virus types 1 and 2. Prog Med Virol 34:89, 1987 150. Arvin AM: Varicella-zoster virus. In Fields BN, Knipe DM, Howley PM, et
al (eds): Fields Virology. Philadelphia: Lippincott-Raven, 1996:2547 151. Hammerschlag MR, Gershon AA, Steinberg SP et al: Herpes zoster in an adult recipient of live attenuated varicella vaccine. J Infect Dis 160:535, 1989 152. Plotkin SA, Starr SE, Connor K et al: Zoster in normal children after varicella vaccine. J Infect Dis 159:1000, 1989 153. Gershon AA: Live attenuated varicella vaccine use in immunocompromised children and
adults. Pediatrics 78:757, 1986 154. Plummer G: Serological comparison of the herpes viruses. Br J Exp Pathol 45:135, 1964 155. Mitchell WJ, Deshmane SL, Dolan A et al: Characterization of herpes simplex virus type 2 transcription during latent
infection of mouse trigeminal ganglia. J Virol 64:5342, 1990 156. Neumann-Haefelin D, Sundmacher R, Wochnik G et al: Herpes simplex virus types 1 and 2 in ocular disease. Arch Ophthalmol 96:64, 1978 157. Rosenwasser GOD, Greene WH: Simultaneous herpes simplex virus type 1 and 2 keratitis in acquired immunodeficiency
syndrome. Am J Ophthalmol 113:102, 1992 158. Ganatra JB, Chandler D, Santos C et al: Viral causes of the acute retinal necrosis syndrome. Am J Ophthalmol 129:166, 2000 159. Van Gelder RN, Willig JL, Holland GN et al: Herpes simplex virus type 2 as a cause of acute retinal necrosis syndrome
in young patients. Ophthalmology 108:869, 2001 160. Itoh N, Matsumura N, Ogi A et al: High prevalence of herpes simplex virus type 2 in acute retinal necrosis
syndrome associated with herpes simplex virus in Japan. Am J Ophthalmol 129:404, 2000 |