TABLE 88-1. Diagnostic Tests for Specific Viruses
| Virus | Cytologic | Culture | Serology | Light Microscopy (Papanicolaou or Giemsa Stain) |
| Herpes simplex virus | IFA | Many animal cell lines; reliable | ELISA, | Multinucleated giant cells |
| DFA | Comp Fix | Eosinophilic intranuclear inclusions (epithelial cells) | ||
| IP | DFA, | |||
| EIA | IFA | |||
| Varicella-zoster viruses | IFA | Human cell lines; fairly reliable | ELISA, | Multinucleated giant cells |
| DFA | IFA | Eosinophilic intranuclear inclusions (epithelial cells) | ||
| Neut Ab, | ||||
| Comp Fix | ||||
| Adenovirus | IFA | Human embryo cell lines; difficult | ELISA, | Eosinophilic intranuclear inclusions (early) |
| DFA | Comp Fix | |||
| EIA | Hem-Inhib | Basophilic intranuclear inclusions (late) (epithelial cells) | ||
| Lymphocytes and degenerating epithelial cells | ||||
| Cytomegalovirus | IFA | Human cell lines; difficult | ELISA | Eosinophilic intranuclear inclusions |
| DFA | Comp Fix (IgG) | Cytomegalic cells | ||
| Ind-Hem | Basophilic cytoplasmic inclusions | |||
| Epstein-Barr | Specific human lines; difficult | ELISA | ||
| Eye disease- may be immunologic | DFA (to capsid antigen) | |||
| ACIF | ||||
| Human immuno-deficiency virus | Specific cell lines; very difficult | ELISA | ||
| LA | ||||
| Western blot (Protein) | ||||
| Enterovirus | IFA | Organ-culture; difficult | ELISA | Predominantly lymphocytes |
| DFA | Neut Ab | Degenerating epithelial cells |
IFA, indirect immunofluorescence; DFA, direct immunofluorescence; IP, immunoperoxidase; EIA, enzyme immunoassay (a form of enzyme-linked immunosorbent assay); ELISA, enzyme-linked immunosorbent assay; Comp Fix, complement fixation; Neut Ab, neutralizing antibodies; Hem-Inhib, hemagglutination inhibition; ACIF, anticomplement immunofluorescence; LA, latex agglutination; Ind-Hem, indirect hemagglutination.
TABLE 88-2. Commonly Used Ocular Diagnostic Techniques
| Virus | Technique | Tissue | Availability | Time to Diagnosis | Relative Cost |
| Herpes simplex virus | Culture | Conjunctiva Cornea | Clinical laboratory | 24–72 hoursa | $$ |
| Aqueous humor | |||||
| DFA/IFA | Conjunctiva Cornea | Specialized/university laboratory | <24 hours | $$ | |
| ELISA (e.g., HERPCHECK, Dupont) | Conjunctiva Cornea | Clinical laboratory | 24 hours | $$ | |
| PCR | Conjunctiva Cornea | Specialized/university laboratory | 24–48 hoursc | $$$ | |
| Aqueous humor | |||||
| Vitreous | |||||
| Varicella-zoster viruses | Cultureb | Cornea | Clinical laboratory | 3–5 days | $$ |
| Vitreous | |||||
| PCR | Cornea | Specialized/university laboratory | 24–48 hoursc | $$$ | |
| Aqueous humor | |||||
| Vitreous | |||||
| Adenovirus | EIA (e.g., Adenoclone,CambridgeBioscience)serology (see Table One) | Conjunctiva | Office laboratory | 1 hour | $ |
| Cytomegalovirus | Clinical laboratory | 24 hourrs | $$ | ||
| PCR | Aqueous humor | Specialized/university laboratory | 24–48 hoursc | $$$ | |
| Vitreous | |||||
| Enterovirus | culture | Conjunctiva | Clinical laboratory | 1–7 days | $$ |
| DFA/IFA | Conjunctiva | Specialized/university laboratory | <24 hours | $$ |
DFA, direct immunofluorescence; IFA, indirect immunofluorescence; ELISA, enzyme-linked immunosorbent assay; PCR, polymerase chain reaction; EIA, enzyme immunoassay.
aTime to earliest viral cytopathic effect.
bCulture for VZV has only 40% to 50% sensitivity.
cTime to diagnosis depends on detection format.
VIRAL CULTURE
Viral species differ in the cell lines in which they can grow, although some viruses can grow in a variety of tissues or cells. Herpes simplex virus (HSV), for example, can be isolated after intracerebral inoculation of suckling mice, from the chorioallantoic membrane of embryonated hen eggs, from mink lung, or from several susceptible cell lines. Conversely, some viruses (e.g., adenovirus) are difficult to isolate and may require specific human cell lines for isolation. Cell culture generally includes essential ingredients for cell growth, serum, antibiotics, and antifungals. Depending on the virus in question and its particular cell culture requirements, a specific cytopathic effect on the cell line is achieved.
Cell culture is the gold standard for confirmation of suspected viral infection. Many authorities believe cultures underestimate the true prevalence of active HSV infection,1–3 as viral cultures are quite specific but may lack sensitivity.4 Unfortunately, viral cultures are relatively expensive, are laborious, and require 1 to 5 days. Consequently, this tool is not available at every hospital and is almost never found in a private office.
CYTOLOGY
Exfoliative Cytology
Conjunctival exfoliative cytology, a well-established technique familiar to all ophthalmologists, is an important if underappreciated diagnostic tool. Gram stain, almost universally available in a hospital setting, is perhaps the prototype for diagnostic exfoliative cytology; however, this technique is designed principally for bacteria and fungi rather than viruses.
Giemsa stain emphasizes cellular morphology and can be useful in diagnosing viral infections. Tzanck smear is another simple, inexpensive preparation used to show viral-induced cellular abnormalities.5 HSV infections often show multinucleated giant cells with either Giemsa stain or Tzanck smear.5 These epithelial cellular aggregates with 3 to 20 nuclei have relatively little cytoplasm and a pale halo between nuclei and cytoplasm. Although somewhat difficult to find on a smear, multinucleated giant cells are diagnostic of infection by either HSV or varicella-zoster viruses (VZV). Less specifically, lymphocytes predominate in smears from superficial viral infections.
Intranuclear viral inclusions may be found with Giemsa stain, with Tzanck smear (with eosin),5 or perhaps better with Papanicolaou stain. HSV and cytomegalovirus (CMV) infections produce intranuclear eosinophilic inclusions (Lipschutz inclusions), but these are difficult to find with any of the three stains. Molluscum contagiosum produces eosinophilic cytoplasmic inclusions (Henderson-Patterson inclusions), especially from expressed lesions, although inclusions are best seen with hematoxylin and eosin staining. Although these exfoliative cytologic techniques are perhaps less sensitive and specific than newer immunologic tools, they are inexpensive and quick and are an essential base for many of the newer methods.
The application of these cytologic techniques is not limited to exfoliative cytology. Cell samples from intraocular fluids or ocular biopsy may be evaluated by any of the previously mentioned cytologic techniques. Exfoliative cytologic samples are usually applied directly to a slide or can be obtained by impression cytology.6 However, intraocular fluids are best handled by centrifugation7 or cytocentrifugation8 before microscopic evaluation. Membrane filter techniques,9,10 which require that the fluid be passed through a filter (e.g., Millipore), trapping solid material, are perhaps superior to both centrifugation and cytocentrifugation.10 This technique is especially valuable with scanty specimens. Cell block techniques can be used on the pellet formed with centrifugation as well as for small fragmented biopsy material.
Electron Microscopy
Electron microscopy with negative staining is relatively quick (2 to 5 hours), but it requires an electron microscope, trained personnel, and a sample with an adequate number of viral particles. It is also less sensitive than viral culture techniques. Occasionally the technique is useful, as illustrated by one report of herpetic uveitis diagnosed by examination of cells in the aqueous humor.11
Immunologic Exfoliative Cytology
Immunocytochemistry techniques have been used for more than 30 years. Indirect and direct immunofluorescent techniques, immunoperoxidase techniques, and most recently immunoenzyme techniques are available.
The indirect immunofluorescent technique is performed by placing cells from the infected area on a clean glass slide. A primary antiserum obtained from an animal (e.g., rabbit) vaccinated against the suspected antigen is then applied. A fluorescein-conjugated, second antiserum from a different species (e.g., goat) is directed against the immunoglobulin type of the primary antiserum (e.g., goat antirabbit immunoglobulin) (Fig. 1). Controls include suspected antigen-bearing cells stained with a negative primary antiserum (or not stained at all), followed by all reagents. Such controls provide a measure of nonspecific fluorescence.12
|
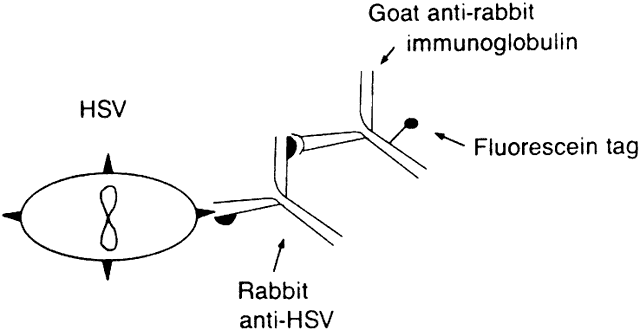
The direct immunofluorescent technique is a variation on the indirect technique.1 Primary antibody is produced by vaccinating an animal (e.g., rabbit) with antigen (e.g., HSV). The primary antibody is then conjugated to fluorescein isothiocyanate. The antiserum containing this conjugated antibody is applied to a slide containing the cells suspected to have the antigen, and the immunofluorescence is evaluated (Fig. 2). The immunofluorescent techniques have advantages and disadvantages. Both the direct and indirect methods are rather sensitive, although generally the indirect test is more sensitive. Slides stained by these techniques can subsequently be stained by conventional methods for light microscopy. These immunofluorescent techniques are particularly good for diagnosing infection by HSV, VZV, CMV, adenovirus, and enterovirus.
|
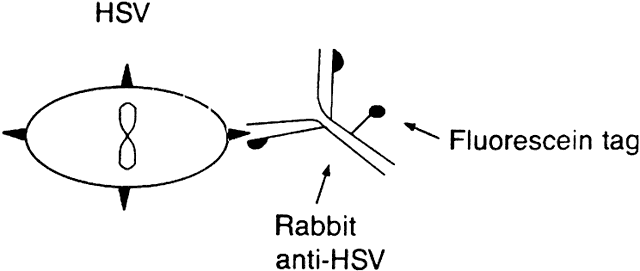
Both indirect and direct techniques may be performed with polyclonal antiserum or monoclonal antibodies. Tests involving polyclonal antibodies may be more sensitive but less specific than tests using monoclonal antibodies. If the antigen to which the monoclonal antibody is directed (e.g., a segment of amino acids from the capsid protein of HSV) is not present on the slide, the antibody will not bind to the agent. Hence, monoclonal antibodies are very specific but may lack sensitivity. Although pooled monoclonal antibodies can be used to increase sensitivity, the immunofluorescent techniques are not commonly used in conjunction with monoclonal antibodies because of this problem.
The anticomplement immunofluorescent technique is another variation on immunofluorescent techniques. Primary antibody prepared in animal against the suspected antigen (e.g., HSV) is applied to the slide after application of the infected cells (e.g., HSV-infected corneal or conjunctival cells). Complement that attaches to the antigen-antibody complex is then added. Fluorescein-tagged antibodies to the C3 component of complement are then added to detect the complement bound by the antigen-antibody complex. This technique offers better specificity than the immunofluorescent techniques because it has less nonspecific staining,13 but it is not widely used.
Based on an enzymatic immunoassay similar in many respects to immunofluorescence, immunoperoxidase studies use the activity of horseradish peroxidase rather than fluorescence as the indicator on primary (direct immunoperoxidase) or secondary (indirect immunoperoxidase) antibodies.14 The principles and methods are otherwise similar to those discussed for immunofluorescent techniques. Antigen-antibody immunoperoxidase complexes are detected colorimetrically after the addition of a specific substrate. Enzymatic cleavage of the substrate causes the cells to change color.15 With immunoperoxidase, a light microscope may be used for detection.16 Immunoperoxidase and immunofluorescence differ little in sensitivity or specificity.
Another variation on the immunofluorescent or immunoperoxidase technique is the immuno-gold silver staining technique, believed by some investigators17 to be more sensitive. In this procedure, the specimen is overlaid with a rabbit anti-HSV antiserum, followed by antirabbit anti-HSV antiserum conjugated with colloidal gold. The presence of virus antigen is then detected by light microscopy as distinct black-staining deposits from silver development.18
Peroxidase-antiperoxidase (PAP) techniques are similar to indirect immunofluorescent techniques but distinct from immunoperoxidase techniques. The suspected cells bearing the antigen are applied to the surface of a slide. The slide is stained with, for example, rabbit antibody to the antigen in question. The slide is then stained with a second species (e.g., swine) to rabbit immunoglobulin. Finally, peroxidase and a rabbit antibody to peroxidase are added to the slide. The peroxidase couples with the rabbit antiperoxidase and the swine antibody couples with the molecules of rabbit globulin-peroxidase, creating a large complex (Fig. 3). The immunoglobulin formed to peroxidase must be from the same species (in this example, rabbit) as the immunoglobulin to the antigen. When a substrate is added to stain the peroxidase complex, the activity of the enzyme on the substrate can be detected microscopically.16,19,20
|
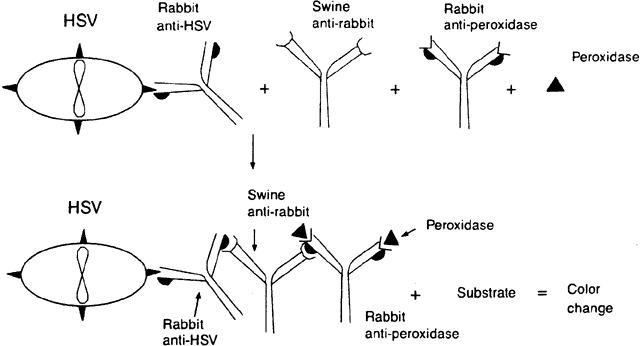
PAP and immunoperoxidase techniques have an advantage over the immunofluorescent techniques. Both peroxidase systems provide a permanent record, whereas immunofluorescent specimens fade with time. Additionally, the PAP method is more sensitive than the immunofluorescent or immunoperoxidase techniques and is sensitive enough to use on paraffin-embedded tissues. However, the technique is time-consuming and tedious.
PAP, immunofluorescent, and immunoperoxidase tests are best used for HSV, VZV, and perhaps CMV antigens. These tests have not been as well evaluated for other viruses.
The avidin-biotinylated peroxidase complex technique begins like the PAP method. The cells containing the suspected antigen are applied to a slide (e.g., HSV-infected epithelial cells). Rabbit anti-HSV immunoglobulin is applied to the slide, and then a biotinylated second antibody to rabbit immunoglobulin (e.g., goat antirabbit immunoglobulin) is applied. Biotinylated horseradish peroxidase and avidin are then applied. Because each avidin molecule has great affinity for biotin and four binding sites for biotin, the avidin will readily attach to the biotinylated peroxidase and biotinylated antibody, creating an amplified sequence and a macromolecular complex (Fig. 4). Once an appropriate substrate is added, a permanent color-positive specimen is achieved.21,22 This complex is believed to amplify binding because of the multiple biotin sites on both the antibody and the peroxidase to which the avidin can attach and because the enzyme remains in place and continues to act on the substrate as long as substrate is present. Specimens containing cells with intrinsic peroxidase (e.g., polymorphonuclear cells have peroxidase) may cause a false-positive result with peroxidase.
|
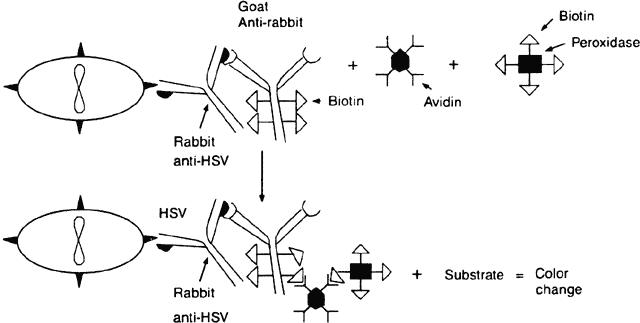
Enzyme-linked immunosorbent assay (ELISA) methods differ from previously mentioned techniques because the antibody is attached to a solid surface such as a microtiter well or plastic bead, and the antibody is said to capture the antigen in the specimen. Alternatively, if desired, the antigen can be attached to the solid phase and used to capture the antibody in the specimen—hence the name immunosorbent. ELISA techniques vary,23–26 but the principles can be illustrated by review of the following example of the direct noncompetitive “sandwich” method. A specimen (e.g., corneal or conjunctival epithelial cells) is inoculated into a plastic microtiter plate in which the wells have been coated with rabbit polyclonal antibodies to the suspected antigen (e.g., HSV). A monoclonal, biotinylated HSV-specific antibody reagent (from a hybridoma) is added. After rinsing, streptavidin-horseradish peroxidase and indicator substrate are added sequentially. The antigen is captured by the solid (plastic) phase antibody that maintains the antigen during the subsequent rinses. The biotinylated HSV-specific monoclonal antibody binds to the stabilized antigen. The streptavidin-horseradish peroxidase complex binds because of avidin's high affinity for biotin. When the indicator substrate is added, peroxidase will produce a colorimetric change, proving the presence of the complex and, hence, the original antigen (Fig. 5). This, too, is an amplification process because each HSV-specific antibody has multiple biotin molecules and the enzyme will continue to act on the substrate molecules present in abundance.
|
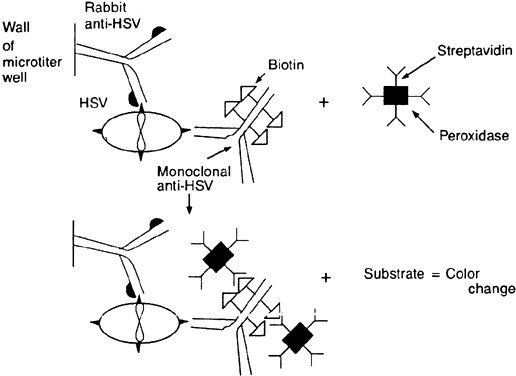
One such commercially available ELISA for HSV, HERPCHEK, has been evaluated, and the results compare favorably with those of culture.27 Importantly, no false-positive results were recognized when patients with other ocular infections, including VZV, were tested.27 Other direct, commercially available ELISAs have shown good sensitivity and specificity in the diagnosis of genital HSV.28
Adenovirus can also be identified by a commercially available direct ELISA (Adenoclone), which uses monoclonal antibodies against the adenovirus capsid hexon. These monoclonal antibodies are fixed in the solid state to the microtiter well. The hexon antigen is common to all known serotypes of adenovirus, so the test may be used for all infections suspected to be caused by adenovirus, and it is simple to perform. It was found to be 77% sensitive (within the first week) and 100% specific.25
ELISA techniques for detection of serum antibodies to VZV, CMV, human immunodeficiency virus (HIV), or other agents are reliable and well accepted. HERPCHEK was shown to be quite sensitive in early trials,27 although tests for nonocular cellular specimens were not as sensitive as viral culture.23 Because ELISA tests will show viral antigen whether viable or not, they may be used when collection techniques are not suitable for cell culture. Additionally, the ELISA tests are relatively inexpensive and yield visually readable results.
MOLECULAR BIOLOGIC TECHNIQUES
The development of nucleic acid hybridization techniques and DNA amplification methods has revolutionized the field of molecular biology and has found numerous applications in viral research. Aside from being a research tool, several techniques, particularly the polymerase chain reaction (PCR), have become vital clinical tools in the diagnosis of viral disease.
Polymerase Chain Reaction
The PCR involves in vitro enzymatic amplification of specific DNA sequences through repeated cycles of oligonucleotide-directed DNA synthesis to amplify a target DNA sequence of low quantity to a detectable level. In several hours, 30 cycles of PCR can amplify a target DNA sequence 1 billionfold. First described in 1985 by workers at the Cetus Corporation,29,30 the technique involves repetitive cycling of three simple reactions: denaturation of native double-stranded DNA, annealing of oligonucleotide primers, and primer extension (Fig. 6). These reactions occur in a single microfuge tube containing 1 ng to 1 mcg of sample DNA and the following reagents:
|
- Each of two oligonucleotide primers (usually 20 to 30 base pairs in length) that
are complementary to sequences flanking the target DNA sequence
- Magnesium-containing reaction buffer
- Excess amounts of the four deoxyribonucleotide triphosphates (A, C, T, G)
- Heat-stable Taq DNA polymerase isolated from the thermophilic bacterium Thermus aquaticus31
- Double-distilled water to bring the final reaction solution volume to 50 or 100 mcl.
Depending on the thermocycler used, mineral oil may be layered over the reaction mixture to prevent evaporation.
DNA from a tissue or cytologic sample must first be prepared using traditional techniques involving phenol/chloroform extraction followed by alcohol purification. Some samples, such as corneal and conjunctival scrapings, can be prepared by placing the specimen in a modified buffer solution containing detergent (e.g., sodium dodecyl sulfate) to lyse cells, followed by heating or boiling of the specimen to denature proteins. Although the extracted DNA is not as pure as that obtained using traditional techniques, this simple method can be used to amplify clinical specimens effectively.
Denaturation of the reaction mixture involves heating to 94°C to separate double-stranded DNA into single strands. The DNA remains in single strands until the temperature is lowered in the second, primer-annealing, step of the PCR. Single-stranded target DNA may then anneal to single-stranded oligonucleotide primers, which are present in huge excess in the reaction mixture. The temperature used for annealing is usually in the range of 37°C to 65°C and is determined empirically along with the duration of the reaction, depending on the stringency required.
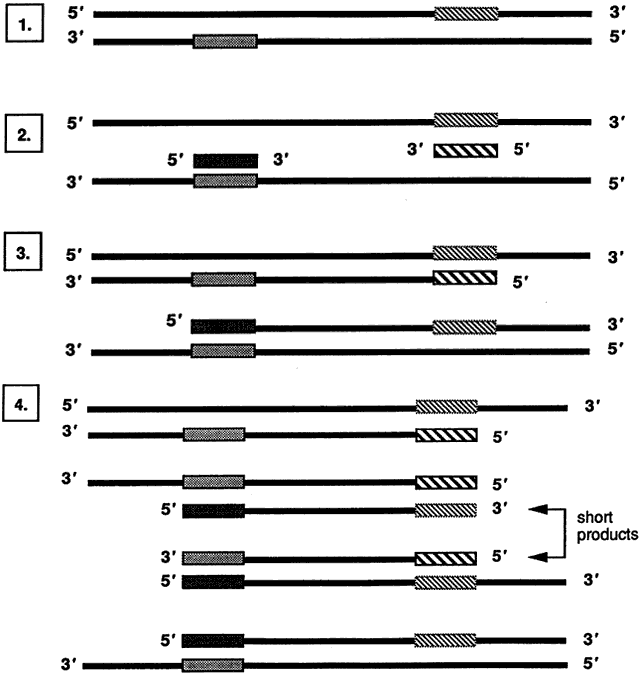
DNA synthesis occurs in the final step of the PCR, whereby annealed primers are extended by Taq DNA polymerase in the presence of excess deoxyribonucleotide triphosphates (dNTPs), usually at a temperature of 72°C. DNA synthesis begins at the 3' end of the primer and proceeds in the 5' to 3' direction. A new complementary strand of DNA is synthesized from each annealed primer. Each newly synthesized strand of DNA can then serve as a template for further primer extension reaction. This results in a geometric amplification of DNA with each cycle of the PCR, hence the aptly named chain reaction that makes this technique exquisitely sensitive in detecting a target DNA sequence.
Most PCRs require at least 30 cycles that progressively deplete the relative numbers of primers and dNTPs while the number of short products expands geometrically (see Fig. 6). The repetitive cycling of temperatures for precise periods of time is conveniently performed in commercially available automated thermocyclers that can process up to 96 samples in a few hours.
PCR products are visualized by electrophoresis of sample aliquots in agarose or polyacrylamide gels, followed by staining with ethidium bromide. The size of amplification products is determined by using molecular weight markers during gel electrophoresis. The identity of amplified products can then be confirmed by Southern blot hybridization using internal DNA oligonucleotide probes, or by sequence analysis of the products.
PCR can also be used to detect RNA sequences and is more sensitive than Northern transfer or in situ hybridization.32 Because DNA polymerase will not work on RNA templates, retroviral reverse transcriptase is used to synthesize complementary DNA (cDNA) to the RNA. This is then amplified by PCR.
The exquisite sensitivity of PCR in amplifying small quantities of target DNA sequences is one of its major drawbacks. False-positive results may occur after inadvertent transfer of a minuscule quantity of DNA into reagents from neighboring sample tubes. To avoid false-positive results, techniques have been developed that include physical separation of PCR reagents and DNA samples,33 and “sterilization” of PCR products against further amplification (e.g., by ultraviolet irradiation).34–37 The former involves preparing samples in a separate room or biosafety hood from that in which DNA samples are stored and amplified. Eppendorf tubes and deionized water used in the procedure should be autoclaved, and gloves should be worn at all times and changed frequently to avoid transfer of amplified DNA. Separating reagents into aliquots and preparing master mixes of reagents also helps prevent contamination by minimizing the number of sample transfers. Eppendorf tubes containing a lyophilized reaction mixture pellet (containing dNTPs, reaction buffer, and Taq DNA polymerase) are now commercially available and require only the addition of primers, DNA sample, and deionized water. Positive displacement pipettes or disposable pipette tips containing plugs prevent cross-contamination from aerosolized DNA in the barrel of a pipette device. DNA should also be the final agent added to any reaction solution. Finally, all PCR runs should contain positive and negative control DNA samples, along with a reagent blank (no added DNA) to help locate contamination (false-positive results), failure or inhibition of the PCR itself (false-negative results), or inadequate nucleic acid extraction or purification from the sample.
Traditionally, detection of amplified PCR products was through gel electrophoresis, often combined with a hybridization process using radioactive probes. Although sensitive, these methods are time-consuming, labor-intensive, and costly, making them poor techniques for the clinical diagnostic laboratory. Nonisotopic detection formats have been developed in which amplified PCR products are detected using an ELISA format, with biotinylated oligonucleotide probes bound to microtiter plates that undergo a colorimetric reaction by means of a streptavidin-horseradish peroxidase conjugate.38,39 This semiautomated technique can provide PCR results in 2 to 4 hours, as opposed to the 12 to 24 hours required with traditional PCR/isotopic hybridization methods. DNA probes to a portion of a viral genome can also be used on nonamplified samples using similar colorimetric detection formats with reasonable sensitivity and specificity.
VIRAL SEROLOGY
Serologic diagnosis of viral infections is usually an adjunctive test for ocular infection. A positive serologic test does not necessarily indicate that the suspect virus is the cause of the ocular infection.
Enzyme-Linked Immunosorbent Assay
A variety of both direct and indirect ELISA techniques are available to detect antibodies (IgG or IgM) to one or more viral pathogens. For example, IgG to VZV can be evaluated as follows. Serially diluted serum is added to microtiter wells coated with viral antigen. Peroxidase-linked rabbit antihuman IgG is added to each well. Appropriate substrate will produce a colorimetric change.40,41
For serologic diagnosis, ELISA methods are more sensitive and less cumbersome than immunofluorescent or immunoperoxidase antibody techniques,40 although the reagents are potentially carcinogenic. IgG and IgM ELISA techniques have been adapted to evaluate infection by Epstein-Barr virus, VZV, CMV, and HSV.42–44 Primary HSV may be diagnosed by a fourfold increase in antibody titer between paired sera drawn at least 2 weeks apart (preferably longer). Serologic testing in recurrent HSV is useless. Recurrent VZV (shingles) may be similarly documented by a fourfold rise in IgG titers. All screening tests for HIV are currently performed using ELISA techniques. These methods are highly sensitive but do give some false-positive results.
Radioimmunoassay
Radioimmunoassay is very similar to ELISA except the final portion of the antihuman immunoglobulin antibody is labeled with a radioactive substance. This technique is quite specific and sensitive but requires the use of radioactive material.
Viral Antibody Neutralization
Most viruses induce antibodies that will neutralize the infectivity of that virus. This principle can be effectively applied to diagnose the presence of viral antibodies (but not necessarily active virus). Serially diluted serum is placed in microtiter wells. Virus is added, and the mixture is then cultured. Cytopathic effect will occur in all wells in which there is not sufficient neutralizing antibody to prevent infectivity of the virus, allowing for direct measurement of the quantity of antibody.
Complement Fixation
Complement fixation is a rather complicated, tedious test, but satisfactory results can be obtained. The technique relies on the immunologic principle that antigen-antibody complexes fix complement. If there is no antibody, complement remains free, and free complement can be made to lyse red blood cells. Viral antigen is added to serially diluted serum, and complement is then added. Sensitized red blood cells (e.g., sheep red blood cells) are added, and any free complement will lyse the cells. The amount of antibody can be determined by the serial dilutions that do not lyse the cells.
Hemagglutination Inhibition Test
Many viruses have the ability to agglutinate the red blood cells of certain species. However, if neutralizing antibodies are present, the process of hemagglutination will be inhibited. Hemagglutination inhibition, then, can be used to assess the quantity of virus and susceptible red blood cells. This test can be standardized to define the quantity of neutralization antibody.
Latex Agglutination
Latex agglutination involves coating latex beads with a viral antigen (e.g., HIV envelope protein DNA) and then exposing the beads to serum suspected to harbor antibodies to the target antigen. Antibodies react with the beads to create a clumping in the suspension, which can be read visually in several minutes.
Viral serology is not as helpful with diagnosis as culture, exfoliative cytology (including immunodiagnostic techniques), or DNA amplification and probing of a tissue specimen. A positive serologic result simply indicates prior exposure to an infectious agent. Nonetheless, serology is often an important adjunct in viral diagnosis.