VIRAL COMPONENTS
A virion is the entire viral infectious unit, including the nucleic acid, the capsid, and if present the envelope. Viral nucleic acids consist of either RNA or DNA. RNA viral genome may be either single- or double-stranded, and in the case of single-stranded viruses, either positive-sense (same polarity as mRNA) or negative-sense (opposite polarity to mRNA). Further, RNA viral genomes are either segmented, with discrete nucleic acid molecules, or nonsegmented, with all the genetic information on a single nucleic acid molecule. Finally, DNA and RNA genomes may be present either in a linear or circular (episomal) form. These characteristics of nucleic acid structure determine in large part the specific mechanics of viral replication.
The viral capsid is the protein shell that surrounds the viral nucleic acid. The capsid interacts internally with the genome to stabilize it, protects the genome from the external environment, and in the case of nonenveloped viruses, expresses on its surface the ligand for virus-host cell binding. The viral capsid proteins also assist in delivery of the viral genome to the intracellular site of viral replication. Thus, viral capsid structure is integrally related to many viral functions, in particular transmission, attachment, and entry into host target cells, but also virion assembly and egress. The capsid and nucleic acid together are referred to as the nucleocapsid. Occasionally, as with herpesviruses, the nucleocapsid is surrounded by an additional protein layer, the tegument.
Capsid structure is specified by the viral genome, and economy of genomic size frequently dictates a capsid of repeating protein subunits. Simplicity further dictates that subunits interact in symmetrical forms with conserved subunit interactions. Common capsid structural motifs include the icosahedron, with its 20 plane surfaces, and the helix.4 Electron microscopy and x-ray diffraction crystallography, in conjunction with nucleic acid and protein sequencing, are the principal techniques applied to delineation of capsid structure.
An envelope surrounds the capsid of some virus families. The envelope consists of viral genome-encoded glycoproteins embedded in a host cellderived lipid bilayer. Viral glycoproteins act as ligands (antigens) for neutralizing antibodies directed against the virus. In the initial stages of infection, envelope glycoproteins mediate attachment of the virus to its receptor on the host cell surface and fusion of the viral envelope with the host cell membrane. During viral replication, viral-encoded glycoproteins are targeted on a molecular level to specific membranes in the host cell to serve as sites of interaction between the viral nucleocapsid and the host cell membrane before budding. Cell membranes used by enveloped viruses include the nuclear envelope, endoplasmic reticulum, Golgi apparatus, and plasma membrane. Polarized epithelial cells, such as those found at mucosal surfaces, maintain tight intercellular junctions and possess biochemically and morphologically distinct apical and basolateral cell membranes. Because of differential targeting of viral glycoprotein into apical versus basolateral membranes, polarized cells typically release enveloped viruses from either the apical or basolateral cell surface. Virus shed apically into mucosal secretions such as the tear film creates the potential for transmission. Virus shed basolaterally may infect deeper tissues or disseminate.5
The viral envelope lipid bilayer is vulnerable to damage by ultraviolet light, detergents, alcohols, and general-use antiseptics. Enveloped viruses such as herpes simplex virus or human immunodeficiency virus are therefore intrinsically susceptible to the external environment. Nonenveloped viruses such as adenoviruses may be quite resistant to degradation, even under relatively harsh conditions.6
VIRAL TROPISMS: RECEPTOR BINDING AND EARLY EVENTS IN INFECTION
Viral tropisms for specific cell types and tissues are not random. Infection depends on the presence on the viral capsid surface (nonenveloped viruses) or envelope (enveloped viruses) of a ligand that can bind to a receptor on the target cell. Viral ligands are typically glycoproteins. Host cell virus receptors are diverse. Although viral ligand-host cell receptor interaction is essential for adsorption of the virus to the cell surface, the ligand receptor complex often also mediates subsequent internalization of the virus and uncoating of the capsid.
The polarized location of the virus receptor on epithelial tissues with distinct apical and basolateral cell surfaces and the changes in receptor expression during cell differentiation largely determine tissue susceptibility to infection in vivo. For example, virus receptor expression only on the basolateral surfaces of less differentiated epithelial cells would permit infection by virus presented across an underlying basement membrane, but not by virus present in mucosal fluids or on undamaged skin.
Viruses presumably evolved the capacity to bind existing constitutive host cell membrane components with essential primary cellular functions (Table 2). Therefore, binding of virus to a cell surface component subverts the natural function of that cellular molecule. For example, the B lymphocyte receptor for Epstein-Barr virus is the C3d complement receptor, CD21.7,8 Rhinoviruses bind to intercellular adhesion molecule-1 (ICAM-1),9–11 present on nasopharyngeal12 and conjunctival13 epithelial cells. Human papillomavirus (HPV) appears to bind the α6 component of the α6β4 integrin complex.14 Adenovirus type 2 uses the CAR protein for attachment15 and an integrin for internalization.16
TABLE 85-2. Selected Ocular Viruses and Their Possible Receptors
| Virus | Host Cell Receptor |
| Herpes simplex virus | Heparan sulfate32,33 |
| Human cytomegalovirus | Heparan sulfate34 |
| Epstein-Barr virus | CD217,8 |
| Human papillomavirus | Integrin α614 |
| Rhinovirus | ICAM-19–11 |
| Influenzavirus | Sialic acid35,36 |
| Vaccinia virus | Epidermal growth factor receptor37 |
In a classic lytic viral infection, virus replication diverts cellular protein production machinery over to the synthesis of viral proteins. However, before shutdown of host macromolecular synthesis, the cell may respond to viral infection by upregulation of specific genes. For instance, binding of cytomegalovirus to cells in vitro stimulates production of proto-oncogenes.17 Adenovirus binding stimulates the rapid induction of host cell-derived proinflammatory cytokines by the Raf/MAPK signal transduction pathway.18 The nonviral cellular function of the host cell virus receptor probably influences the initial molecular response to infection.
FUNDAMENTALS OF VIRAL INFECTION, REPLICATION, AND PATHOGENESIS
Viruses may infect the human host via the placenta and birth canal, through ingestion of breast milk, by inhalation of airborne secretions, through contaminated food, by insect bite or inadvertent passage with intravascular injections, and by intimate or sexual contact. Ocular infection by viruses most often follows direct contact with virus externally from infected secretions in the birth canal (herpes simplex virus, HPV), on fomites (adenovirus), or airborne particles (rhinovirus), or is acquired during viremia (human cytomegalovirus, measles virus). Other mechanisms of ocular viral infection include extension from contiguous adnexal disease (herpes simplex virus), spread from the upper respiratory tract via the nasolacrimal duct (rhinovirus), and transplacental passage of infectious virus (rubella virus). Rarely, ocular infection may disseminate elsewhere (enterovirus 70).19 A classification of viruses infecting the human eye and adnexa is presented in Table 3.
TABLE 85-3. Classification of Viruses Affecting the Human Eye
| Virus | Family | Subfamily/ Genus | Nuc. Acid | Env. | Capsid | Ocular Target |
| Herpes simplex virus, type 1(HHV1) | Herpesviridae | Alphaherpesvirinae/ Simplexvirus | dsDNA | + | Icosahedral | Eyelid |
| Conjunctiva | ||||||
| Cornea | ||||||
| Trabecular meshwork | ||||||
| Uvea | ||||||
| Retina | ||||||
| Herpes simplex virus, type 2 (HHV2) | Herpesviridae | Alphaherpesvirinae/ Simplexvirus | dsDNA | + | Icosahedral | Eyelid |
| Conjunctiva | ||||||
| Cornea | ||||||
| Trabecular meshwork | ||||||
| Uvea | ||||||
| Retina | ||||||
| Varicella zoster virus (HHV3) | Herpesviridae | Alphaherpesvirinae/ Varicellovirus | dsDNA | + | Icosahedral | Eyelid |
| Conjunctiva | ||||||
| Cornea | ||||||
| Trabecular meshwork | ||||||
| Uvea | ||||||
| Retina | ||||||
| Optic nerve | ||||||
| Epstein-Barr virus (HHV-4) | Herpesviridae | Gammaherpesvirinae /Lymphocryptovirus | dsDNA | + | Icosahedral | Lacrimal gland |
| Conjunctiva | ||||||
| Cornea | ||||||
| Uvea | ||||||
| Retina | ||||||
| Optic nerve | ||||||
| Human cytomegalovirus (HHV5) | Herpesviridae | Betaherpesvirinae/ Cytomegalovirus | dsDNA | + | Icosahedral | Retina |
| Optic nerve | ||||||
| Human herpes virus 6 (HHV6) | Herpesviridae | Betaherpesvirinae/ Roseolovirus | dsDNA | + | Icosahedral | Retina |
| Human herpes virus 7 (HHV7) | Herpesviridae | Betaherpesvirinae/ Roseolovirus | dsDNA | + | Icosahedral | ? |
| Human herpes virus 8 (HHV8) | Herpesviridae | Gammaherpesvirinae | dsDNA | + | Icosahedral | Conjunctiva (Kaposi sarcoma) |
| Adenovirus | Adenoviridae | Mastadenovirus | dsDNA | - | Icosahedral | Conjunctiva |
| Cornea | ||||||
| Human papillomavirus | Papovaviridae | Papillomavirus | dsDNA | - | Icosahedral | Eyelid |
| Conjunctiva | ||||||
| Cornea | ||||||
| Smallpox (variola) virus | Poxviridae | Orthopoxvirus | dsDNA | + | Complex | Eyelid |
| Conjunctiva | ||||||
| Cornea | ||||||
| Uvea | ||||||
| Optic nerve | ||||||
| Vaccinia virus | Poxviridae | Orthopoxvirus | dsDNA | + | Complex | Eyelid |
| Conjunctiva | ||||||
| Cornea | ||||||
| Molluscum contagiosum virus | Poxviridae | Molluscipoxvirus | dsDNA | + | Complex | Eyelid |
| Conjunctiva | ||||||
| Cornea | ||||||
| Orf virus | Poxviridae | Parapoxvirus | dsDNA | + | Complex | Eyelid |
| Enterovirus(es): | Picornaviridae | Enterovirus | ssRNA (+) | - | Icosahedral | Conjunctiva |
| (includes poliovirus, coxsackievirus, echovirus, enterovirus) | Cornea | |||||
| Rhinovirus | Picornaviridae | Rhinovirus | ssRNA (+) | - | Icosahedral | Conjunctiva |
| Rubella virus | Togaviridae | Rubrivirus | ssRNA (+) | + | Icosahedral | Cornea |
| Uvea | ||||||
| Lens | ||||||
| Trabecular meshwork | ||||||
| Retina | ||||||
| Globe | ||||||
| Alphavirus/flavivirus: | Togaviridae | Rubrivirus | ssRNA (+) | + | Icosahedral | Conjunctiva |
| (encephalitis, encephalomyelitis, yellow fever, dengue viruses) | ||||||
| Influenzavirus | Orthomyxoviridae | Influenzavirus (A, B, C) | ssRNA (-) | + | Helical | Lacrimal gland |
| Conjunctiva | ||||||
| Episclera | ||||||
| Cornea | ||||||
| Uvea | ||||||
| Retina | ||||||
| Optic nerve | ||||||
| Cranial nerves | ||||||
| Human coronavirus | Coronaviridae | Coronavirus | ssRNA (+) | + | Helical | Conjunctiva |
| Newcastle disease virus | Paramyxoviridae | Paramyxovirus | ssRNA (-) | + | Helical | Conjunctiva |
| Cornea | ||||||
| Parainfluenzavirus(es) | Paramyxoviridae | Paramyxovirus | ssRNA (-) | + | Helical | Conjunctiva |
| Respiratory syncytial virus | Paramyxoviridae | Pneumovirus | ssRNA (-) | + | Helical | Conjunctiva |
| Mumps virus | Paramyxoviridae | Paramyxovirus | ssRNA (-) | + | Helical | Lacrimal gland |
| Conjunctiva | ||||||
| Sclera | ||||||
| Cornea | ||||||
| Uvea | ||||||
| Optic nerve | ||||||
| Cranial nerves | ||||||
| Measles (rubeola) virus | Paramyxoviridae | Morbillivirus | ssRNA (-) | + | Helical | Conjunctiva |
| Cornea | ||||||
| Uvea | ||||||
| Retina | ||||||
| Optic nerve | ||||||
| Cranial nerves | ||||||
| Rift Valley fever virus | Bunyaviridae | Bunyavirus | ssRNA (-) | + | Coiled | Retina |
| Colorado tick fever virus | Reoviridae | Coltivirus | dsRNA (+/-) | - | Icosahedral | (?: reported to cause photo- phobia, retro-ocular pain) |
| Rabies virus | Rhabdoviridae | Lyssavirus | ssRNA (-) | + | Helical | (Transmission via corneal button) |
| Human immunodeficiency virus | Retroviridae | Lentivirus | ssRNA (+) | + | Coiled | Lacrimal gland |
| Retina |
+, Enveloped;-, nonenveloped; (+), positive-sense RNA genome; (-), negative-sense RNA genome.
The objective of infection for a virus is the generation of viral progeny. The synthesis of viral-encoded proteins is essential to the ability of the virus to replicate and determines the effect of viral infection on the cell. Although differences exist between enveloped and nonenveloped viruses in the mechanics of infection, the replicative cycle of all viruses can be summarized in six stages (Fig. 1):
|
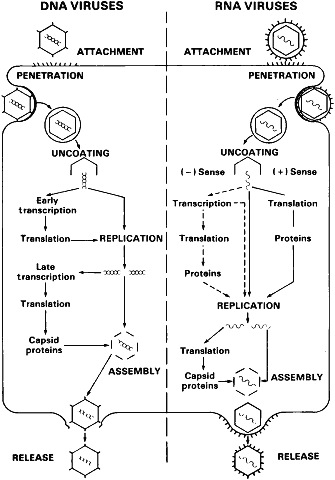
- Attachment
- Penetration
- Uncoating
- Replication
- Assembly
- Release.
Host cell receptors may be protein, glycoprotein, lipid, or carbohydrate.20 After adsorption, penetration occurs by endocytosis or translocation, or in the case of enveloped viruses, fusion of the envelope with the host plasma membrane. Virus capsid components play an active role in transport of the virus into the cell. Uncoating, or shedding of capsid components, typically occurs in the cell cytoplasm. Replication takes place in the nucleus for most DNA viruses and in the cytoplasm for most RNA viruses. Mechanisms of viral replication are summarized in Figure 2. Assembly of the virus, the process by which capsid is added to newly replicated genome, typically occurs in the cytoplasm. Release occurs by budding or lysis.
|
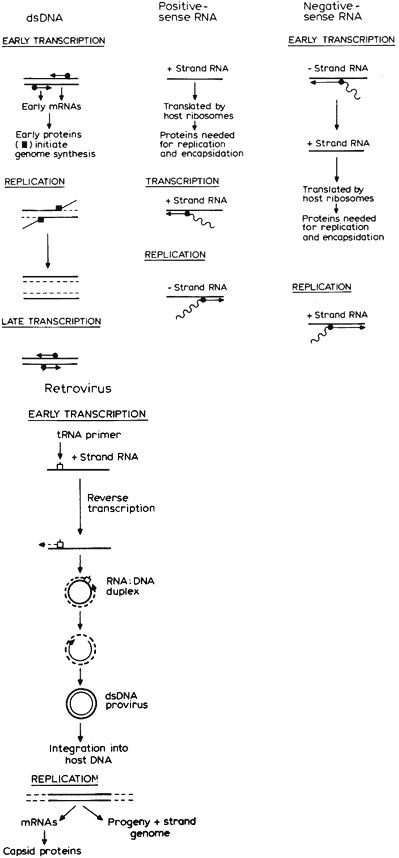
Transcription of viral nucleic acid to produce the enzymatic and structural proteins necessary for replication varies with the type of viral genome. With the exception of the positive-sense single-stranded picornaviruses, alphaviruses, and flaviviruses, it is necessary first to transcribe an mRNA. DNA viruses that replicate in the cell nucleus can use cell-derived polymerases; otherwise, generation of a viral-encoded RNA polymerase is required. Lastly, because eukaryotic host cells do not recognize internal initiation sites within mRNA molecules, posttranslational modifications of viral proteins by cellular or viral enzymes are often used to produce the individual proteins necessary for replication and maturation.
Assembly of infectious virus and subsequent release of virus from the cell are tightly linked processes and largely determine the outcome of infection. The assembly of nonenveloped viruses in the cell nucleus or cytoplasm typically exposes the cell to capsid components that may inhibit cellular function and cause cell death. To acquire envelopes, viruses encode proteins for insertion into host cell membranes that then act as binding targets for immature virions. Egress of the virus via budding sometimes leads to cell lysis (herpesviruses).
Viruses cause disease by a broad array of mechanisms, including altered cellular metabolism caused by viral gene products, altered host gene expression mediated by interactions between viral proteins and the host genome, and host immune response to viral infection of the cell. The end results of viral infection may be frank destruction of host tissues; disrupted function on cellular, tissue, organ, or systemic levels; recurrent disease from intermittent viral expression over time; neoplastic transformation; or immunologically mediated disease.
VIRAL IMMUNOPATHOGENESIS
Virus infections are limited by natural killer cells, B lymphocyte-derived antibodies, and effector T lymphocytes. Unlike B and T lymphocytes, natural killer cells act without antigen specificity or immunologic memory. Interferon-stimulated natural killer cells limit the extent of viral infection early on, before the machinery of acquired antigen-specific immunity has fully engaged. Additionally, activated natural killer cells attack cells with reduced MHC class 1 expression to counter viral evasion of MHC class 1 presentation (see below).
Antibodies neutralize free virus in blood or mucosal secretions. They also mediate cell death of infected cells through complement-mediated killing and by antibody-dependent cell-mediated cytotoxicity. Viral neutralization by antibody depends on recognition of viral epitopes present on virus surfaces such as envelope glycoproteins or, in the case of nonenveloped virus, surface capsid proteins. High quantities of virus-antibody immune complexes in the blood can induce immune complex-mediated immunopathology at distant tissue sites.
CD8+ cytotoxic T lymphocytes (CTLs) typically recognize viral epitopes in the context of MHC class 1 molecules expressed on the surface of virusinfected cells and are critical to the elimination of virus-infected cells. All nucleated cells express class 1 molecules, so any virus-infected cell may be a target for CTLs. Killing occurs through a directional release of perforin and granzymes with a minimum of bystander damage. However, stimulation of T-cell immunity may be accompanied by production of tumor necrosis factor and other cytokines that have deleterious effects at local and systemic levels.
Some viruses possess the means to evade the host's immune system. A herpes simplex virusencoded protein, ICP47, successfully competes with antigenic viral peptides for transport into the endoplasmic reticulum, where peptides are loaded onto the MHC complex.21 Thus, herpes simplex virus-infected cells can be resistant to CTL lysis. Similarly, human cytomegalovirus-encoded US11 dislocates MHC molecules into the cytosol, where they are degraded by cell proteases.22 These and other examples of the means by which ocular viruses may evade the immune system are presented in Table 4.
TABLE 85-4. Ocular Viruses and Molecular Means by Which They Evade Host
Immunity
| Virus | Examples of Immune Escape Mechanisms |
| Herpes simplex virus | Virus-encoded ICP47 blocks peptide translocation to MHC class 121 |
| Virus-encoded proteins bind and neutralize complement components38 | |
| Latency in sensory neurons | |
| Human cytomegalovirus | Virus-encoded US11 causes cytosolic degradation of MHC class 1 heavy chains22 |
| Viral MHC class I homologue inhibits natural killer cell attack39 | |
| Latency in glandular tissue | |
| Epstein-Barr virus | Exclusive EBNA-1 expression in type 1 latency reduces recognition by cytotoxic T lymphocytes40 |
| Adenovirus type 2 | Prevents MHC class I transport to cell surface41 |
| Protection from tumor necrosis factor-mediated cytolysis42 | |
| Influenzavirus | Inhibits cytolysis by interferon43 |
| Antigenic shift and drift | |
| Vaccinia virus | Blocks antiviral effect of interferon44 |
Certain viruses produce homologues of human proteins that can influence host immunity. Epstein-Barr virus encodes a homologue of human IL-10.23 Expression of viral IL-10 by infected cells inhibits interferon-γ production and T-cell immunity and results in enhanced survival of virus-infected cells. Human herpesvirus 8 encodes a structural homologue of IL-6, suggested to influence the pathogenesis of Kaposi sarcoma.24 Human cytomegalovirus encodes a homologue for a human chemokine receptor, providing the capacity to divert host cell-derived chemokines and thereby prevent inflammation and viral clearance.25
Viral infections have been implicated in autoimmune disease induction by several mechanisms.26 Some viruses stimulate polyclonal B-cell activation and lead to excessive deposition of immune complexes in sensitive tissues. The altered cytokine milieu associated with viral infection can stimulate autoreactive T cells, resulting in inadvertent damage to normal tissues. Inflammation of immunologically sequestered tissues, such as those in the central nervous system or the eye in the mature adult, could expose previously hidden epitopes and lead to local hyperimmunologic responsiveness, with devastating functional consequences. Finally, shared antigenic determinants between virus and host can lead in genetically susceptible persons to immunologic recognition of self-epitopes (molecular mimicry),27 with immune-mediated damage at distant and ostensibly normal sites.
Viruses that infect the eye or its adnexa produce stereotypic pathologic changes in ocular target tissues. Infection of the eyelid skin by viruses typically induces the formation of vesicles and ulceration. Infection of the conjunctiva results in increased numbers of conjunctival lymphoid follicles and enlargement of the corresponding draining lymph nodes. Although clinical teaching traditionally emphasizes the disparity in appearance of epithelial keratitis with different viral etiologies, viral infection of the corneal epithelium invariably causes punctate epithelial cytopathic effect, evident biomicroscopically as isolated swollen epithelial cells (punctate epithelial keratitis) and loss of individual epithelial cells (punctate epithelial erosions). When extensive, the punctate erosions may coalesce to form confluent epithelial ulcers (dendritic, dendritiform, and geographic ulcers).28 Corneal stromal infection results in white blood cell recruitment to the site of infection,29 with resultant stromal infiltration. Retinal infection leads to retinal necrosis. Viral encephalitis, encephalomyelitis, and meningitis may lead to cranial nerve inflammation and dysfunction of vision or extraocular motility.
OUTCOMES OF VIRAL EYE INFECTION: LATENCY, CARCINOGENESIS, LOSS OF FUNCTION
Although many viruses cause self-limited infections with complete clearance of the virus, some persist indefinitely in the host. For example, adenoviruses persist within nasopharyngeal lymphoid tissue, Epstein-Barr virus in nasopharyngeal epithelial cells and B lymphocytes, herpes simplex and varicella zoster viruses in sensory ganglia, and HPV in skin and mucosal epithelia. Persistent infections may consistently or intermittently produce infectious virus. When not productive of infectious virus, the infection is said to be latent. In latent infections, only limited viral gene expression occurs. Interestingly, latent infection frequently occurs in cell types poorly permissive for lytic infection by the virus. Such is the case with sensory neurons and herpes simplex virus, B lymphocytes and Epstein-Barr virus, and basal skin epithelial cells and HPV. Latent infections tend to occur in slow-cycling cells. HPV and Epstein-Barr virus both persist as circular episomes within the cell nucleus andcan replicate as necessary to maintain themselves on cell division.
Persistent viral infection of susceptible cells can lead to malignant transformation. Viral proteins, whether directly through interaction with the host genome or by interaction with cellular proteins, can induce transformation of the cell and loss of senescence. Perhaps the most elegant example of tumor induction is by HPV.30 HPV tropisms for skin and mucosa derive in part from tissue-specific gene expression.31 HPV types 6 and 11 are maintained in a latent state within basal epithelial cells as circular episomes with very limited viral gene transcription and low copy number. Early viral gene products stimulate cell growth and lead to a skin wart or a conjunctival papilloma. As basal epithelial cells containing HPV mature and differentiate into superficial epithelial cells, they become permissive for complete viral gene expression and produce infectious virus. Neoplastic transformation from HPV type 6 or 11 is rare. In contrast, HPV types 16 and 18 stereotypically integrate their viral genome into host chromosomal DNA, and this in turn has been strongly associated with malignant transformation and squamous cell carcinoma. In the episomal state, transcription of HPV proto-oncogenes E6 and E7 is effectively repressed by the HPV E2 gene product. When HPV genome integrates into host cell chromosomal DNA, the circular (episomal) viral DNA molecule breaks at a recombination site within the E2 open reading frame, resulting in a truncated E2 protein and disinhibition of E6 and E7 transcription. The E6 protein binds to and initiates the degradation of cellular p53 tumor suppressor gene. The E7 protein displaces cellular pRB from its complex with cellular E2F transcription factor. E2F then activates transcription of genes that initiate the cell cycle. Hence, increased cellular levels of E6 and E7 proteins contribute to the malignant phenotype of squamous epithelium infected with HPV types 16 and 18.
The results of viral infection depend on a complicated array of factors. The presence of viral receptors on host cells at a surface exposed to infectious virus, the permissiveness of the cell to the viral gene cycle, the capacity of the host to eliminate the virus balanced against the damage to host tissue caused by the immune response, and finally the fine function of the tissue all determine the functional and anatomic derangements associated with viral infection. For a virus like herpes simplex, tropic for almost all ocular tissues, the outcome of ocular infection varies with the tissue infected. Herpes simplex virus infection of the conjunctiva is self-limited and leaves no visual deficit, whereas infection of the corneal stroma may result in varying degrees of vision loss, and infection of the retina may result in complete loss of useful vision. In contrast, HPV ocular tropism is limited to the conjunctiva, limbus, and eyelid skin. Blinding sequela of HPV infection occurs with malignant transformation of infected tissues.
As classification of viruses proceeds on a molecular genetic level, the mechanisms by which viruses infect ocular cells, destroy critical ocular structures, evade the immune system, and induce cancer will be better understood.