TABLE TWO. Antiprotozoal Drugs for Acanthamoebic Keratitis
| Drug | Formula | Dosage |
| Neomycin | C23H46N6O13 | 1.75–3.5 mg/ml (in commercial solution) or 5–10 mg/ml, topically |
| Paromomycin (aminosidine) | C23H45N5O14 | 10 mg/ml, topically |
| Miconazole | C18H14Cl4N2O | 10 mg/ml, topically |
| Clotrimazole | C22H17ClN4 | 10 mg/ml, topically |
| Ketoconazole | C26H28Cl2N4O4 | 400 mg/day, orally |
| Itraconazole | C35H38Cl2N8O4 | 200 mg/day, orally |
| Propamidine | C17H20N4O2 | 1 mg/ml, topically |
| Hexamidine | C20H26N4O2 | 1 mg/ml, topically |
| Polyhexanide (polyhexamethylene biguanide) | C8H17N5On | 0.2 mg/ml, topically |
| Chlorhexidine | C22H30Cl2N10 | 0.2 mg/ml, topically |
AMINOGLYCOSIDES: NEOMYCIN AND PAROMOMYCIN
Historical Development
The term antibiotic was coined in the 1940s by Waksman to describe chemical substances of microbial origin that inhibit the growth or the metabolic activities of bacteria and other microorganisms. By screening a large number of soil actinomycetes, his laboratory discovered streptomycin, for which the 1952 Nobel Prize in Medicine or Physiology was subsequently awarded. In 1949, Waksman's laboratory found that Streptomyces fradiae produced a group of antibacterial compounds collectively called neomycin.11
Neomycin-containing products are composed largely of neomycin B. Oral neomycin is poorly absorbed and was formerly used to suppress intestinal flora before bowel surgery and to sequester bile acids in treating hypercholesterolemia. Ototoxic and renal side effects occur, and this systemic toxicity has limited neomycin's use to topical dermatologic and ophthalmologic preparations.12 Neomycin was one of the first compounds found to have in vitro amebicidal and cysticidal activity against ocular isolates of Acanthamoeba.13 Neomycin was one of several topical drugs used in treating some of the first reported cases of Acanthamoeba keratitis.14,15
Paromomycin (“closely resembling neomycin”) was isolated from Streptomyces rimosus in 1956 by researchers at Parke-Davis16 and is also produced by S. chrestomyceticus. This oral aminoglycoside was first used in the treatment of intestinal amebiasis and tapeworm infestation and more recently in suppressing cryptosporidiosis. Paromomycin (aminosidine in the United Kingdom) was one of the first compounds found to inhibit acanthamoebae in vitro17 and is possibly more active than neomycin.18 Based on its activity against ocular isolates, paromomycin 1% solution was used in the first patient treated for Acanthamoeba keratitis.19 The amebicidal role of newer aminoglycosides such as geneticin has not been studied.
Chemistry and Preparations
Like other aminoglycosides, neomycin is a highly polar cation that is very stable at room temperature. Neomycin B contains three amino sugars that are linked to an aminocyclitol ring, 2-deoxystreptamine (Fig. 1). Neomycin sulfate is available in several ophthalmic eyedrops and ointments, often in combination with other antibiotics and/or corticosteroids. Neomycin-containing eyedrops are commercially available in concentrations of 1.75 mg/ml in combination with polymyxin B and gramicidin (Neosporin and others), 3.5 mg/ml in combination with polymyxin B (Statrol), 3.5 mg/ml in combination with polymyxin B and a corticosteroid (Cortisporin, Maxitrol, Poly-Pred, and others), and 3.5 mg/ml in combination with a corticosteroid (NeoDecadron). The solid drug (available as 500-mg tablets) or a concentrated solution (available in ampules for urinary bladder irrigation at a concentration of 40 mg/ml in combination with polymyxin B) has been diluted in buffered saline or artificial tears to a concentration of 5 to 10 mg/ml for topical use.
|
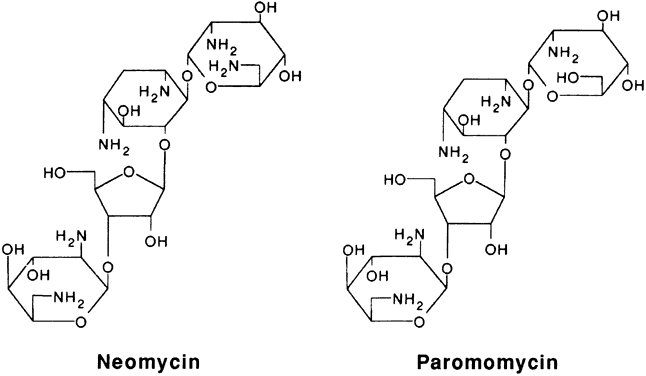
Like neomycin B, paromomycin I is made up of three amino sugars attached by a glycosidic linkage to a hexose nucleus. The drug is water-soluble, and a topical preparation can be prepared by dissolving the powder of the 250-mg oral capsule (Humatin) in isotonic saline or artificial tears to yield a concentration of 10 mg/ml.
Pharmacologic Actions
The aminoglycosides exert antibacterial effects by binding to ribosomes, thereby inhibiting protein synthesis, and perhaps by a direct bactericidal action on the cell membrane. The mechanism of action against Acanthamoeba has not been adequately studied and remains speculative. The aminoglycosides have good amebicidal activity against trophozoites but have limited cysticidal effects for most clinical isolates (Table 3).
TABLE THREE. Average Susceptibility of Most Acanthamoeba Corneal Isolates
| Drug | Minimum Trophozoiticidal Concentration | Minimum Cysticidal Concentration |
| Neomycin | 5–20 μg/ml | 100–1000 μg/ml |
| Paromomycin | 1–5 μg/ml | 100–1000 μg/ml |
| Miconazole | 125–250 μg/ml | 250–1000 μg/ml |
| Clotrimazole | 125–250 μg/ml | 100–1000 μg/ml |
| Ketoconazole | 125–250 μg/ml | 100–1000 μg/ml |
| Itraconazole | 125–250 μg/ml | 100–1000 μg/ml |
| Propamidine | 0.5–1 μg/ml | 5–500 μg/ml |
| Hexamidine | 0.5–1 μg/ml | 5–500 μg/ml |
| Polyhexanide | 0.5–1 μg/ml | 1–5 μg/ml |
| Chlorhexidine | 0.5–1 μg/ml | 1–5 μg/ml |
Compiled from multiple sources.
Neomycin presumably passes through the cytoplasmic membrane of trophozoites, probably by an active aerobic process. By itself, neomycin has a limited effect against cysts. Amebicidal and cysticidal concentrations are typically much higher than corresponding minimal inhibitory concentrations.20 The addition of an agent that disrupts the cyst wall may facilitate a cysticidal or cystistatic effect of neomycin.
Paromomycin can interfere with ribosomal protein synthesis in several microorganisms, but its action on acanthamoebae has not been studied. Cysticidal activity may be related to an effect on membrane permeability.
Dosage and Pharmacokinetics
The oral aminoglycosides are poorly absorbed and must be applied topically for treating acanthamoebic keratitis. One drop every 1 to 2 hours is usually begun during initial therapy. Corneal levels achieved by topical neomycin or paromomycin during inflammation have not been measured.
Adverse Effects
Topical neomycin produces contact hypersensitivity in 5% to 10% of patients. Eyelid edema, conjunctivitis, and punctate corneal erosions may be due to allergic and toxic effects of topical neomycin. While the corneal epithelium is minimally affected by a low dose of topical neomycin,21–23 cytotoxicity occurs at concentrations greater than 5 mg/ml.24 Corneal sensation can be altered with very high levels of neomycin eyedrops.25
The effects of topical paromomycin on the ocular surface have not been adequately studied. Frequent dosing and prolonged administration presumably can slow wound healing and may contribute to conjunctival hyperemia and punctate corneal erosions.
Clinical Experience
Neomycin or paromomycin has been a part of the medical regimen in several cases of Acanthamoeba keratitis,26 but it is difficult to ascertain their clinical value. Permanent quiescence has occurred with combination therapy that includes one of these topical aminoglycosides, but other cases have progressed. The role of neomycin and other antibacterial agents in eradicating or preventing concomitant bacterial growth that might complicate Acanthamoeba corneal infection has not been determined. Whether both aminoglycosides would be additive when used together is not known.
DIAMINIDES: PROPAMIDINE AND HEXAMIDINE
Historical Development
Propamidine was synthesized in 1941 and reported to have antimicrobial properties in 1951. The finding that hydroxystilbamidine was effective in vitro against strains of acanthamoebae led to the discovery that other diamidine congeners might be clinically useful. The addition of propamidine (prop[ane] + amidine) to the medical regimen for Acanthamoeba keratitis seemed to increase the likelihood of successful treatment.27,28 Other diamidines, such as hexamidine (hex[ane] + amidine)29,30 and pentamidine,31 have also been assessed.
Chemistry and Preparations
Propamidine (4,4'-[trimethylenedioxy]dibenzamidine) and hexamidine (4,4'-[hexamethylenedioxy]dibenzamidine) are available as diisethionate salts in 0.1% ophthalmic solutions that are licensed as ocular disinfectants for nonprescription sale as Brolene ( Rhône-Poulenc) in the United Kingdom and as Désomédine (Chauvin) in France, respectively (Fig. 2). Dibromopropamidine ointment (Brolene) is marketed in Great Britain for human and veterinary use.
|
Pharmacologic Actions
The protanated amidine groups interact with the plasma membrane of trophozoites, resulting in cytoplasmic leakage. The molecule must pass through the double wall of cysts, which occurs even though it is composed mainly of hydrophilic chitin. The amebicidal and cysticidal effects partially depend on the lipophilic property of the drug. Diamidines also produce an inhibitory effect by gaining entry into the cell, where the drug blocks molecular linkages and dissociates nucleic acids from proteins. Diamidines can bind to DNA sequences composed of at least four consecutive adenine-thymidine base pairs, although the relationship between nucleotide binding and amebicidal activity is uncertain.32 Aromatic diamidines also inhibit amebic adenosylmethionine decarboxylase.33
There is an additive cysticidal effect between propamidine and other effective agents such as neomycin or paromomycin. In vitro studies show variable cysticidal activity.34,35 Diamidines have little or no effect on microsporidia.
Dosage and Pharmacokinetics
A concentration of 0.1% is commercially available in over-the-counter solutions of propamidine and hexamidine, but the optimal dosage is not known. Treatment is usually begun as one drop every 1 to 2 hours, although an initial frequency of every 15 to 30 minutes has been recommended.36,37
Adverse Effects
Topical diamidines can produce stinging and burning immediately after application. Conjunctival hyperemia and punctate corneal erosions have been attributed to its use.38 Mast-cell degranulation with histamine release is produced by propamidine. Contact hypersensitivity has been described.
Clinical Experience
A multiple-drug regimen is commonly used in the treatment of Acanthamoeba keratitis. While medical control was achieved in some of the early cases, the addition of a diamidine such as propamidine has improved the chance for successful resolution of infection.35 Resistant cases have been encountered,39 and propamidine resistance can emerge during therapy.40
BIGUANIDES: POLYHEXANIDE AND CHLORHEXIDINE
Historical Development
The biguanides were first synthesized in the 1940s in the search for antimalarial agents. The antimalarial biguanides (e.g., chlorguanide) and other guanidine derivatives (e.g., guanethidine) have minimal or inconsistent antiamebic activity, but their discovery led to biguanide compounds with broader antimicrobial properties.
Chlorhexidine was developed by Imperial Chemical Industries in the 1950s.41 When the bis-biguanides such as chlorhexidine were found to be more microbicidal than the monomeric biguanides, polymeric biguanides were developed. Polyhexamethylene biguanide (polyhexanide, PHMB) was developed during the 1960s in several molecular weights.
As a result of the public concern about amebic meningoencephalitis, especially in Australia and New Zealand in the 1970s, an agent was sought to kill free-living amebae in swimming pools and hot tubs. PHMB was found to be amebicidal,42 and this cationic agent was developed as an alternative to chlorination for water sanitation. The proposal that biguanide biocides may play a role in the treatment of Acanthamoeba keratitis was based on in vitro studies at Bath, England. Choosing an empiric concentration of 0.02% (200 μg/ml) that would be expected to give corneal levels of polyhexamethylene biguanide greater than the minimum cysticidal concentration, this diluted swimming pool disinfectant was first successfully used to treat patients responding poorly to conventional antiamebic therapy.43 Further experience showed that it was useful as part of primary treatment.44,45
Chlorhexidine was known to have in vitro efficacy against pathogenic amebae in the 1970s.46 Further laboratory testing confirmed that PHMB, chlorhexidine, and other biguanides (e.g., alexidine) were effective against Acanthamoeba.47 Biguanides emerged as clinically useful agents during the 1990s.
Chemistry and Preparations
Polyhexamethylene biguanide is not commercially marketed as a human medication. It is available in a number of products under different trade names (Baquacil, Cosmocil, Vantocil) as heterodisperse mixtures with differing average molecular weights. The optimal antiamebic concentration for ocular use has not been determined. A 0.00005% concentration of PHMB in a contact lens disinfectant (ReNu) is too low for cysticidal activity.48,49 Serial dilution of a swimming pool disinfectant, containing 20% Baquacil, to a concentration of 0.02% has been successfully used in treating Acanthamoeba keratitis. Concentrations of more than 0.5% are toxic to the ocular surface. Commercial preparations of PHMB may be composed of a mixture of various lengths of polymer chains (Fig. 3). While several hundred repeating units are feasible, most products contain about 5 to 15 hexamethylene units. The degree of polymerization may relate to microbicidal activity,50 and there may be therapeutic differences between Baquacil, Cosmocil, and other polyhexanide preparations.
|
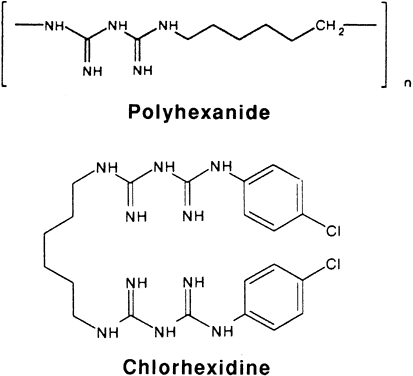
Chlorhexidine (1,6-di[4-chlorophenyl-diguanidino]hexane) digluconate is marketed as a skin disinfectant (Hibitane) in a detergent or alcohol base (e.g., Hibiclens and Hibistat) and for use as a mucosal rinse (e.g., Peridex).51 A concentration of 0.02% in isotonic saline or artificial tears has been successfully used in treating Acanthamoeba keratitis.52
Biguanide solutions can become contaminated. The use of preservatives may be considered when making extemporaneous dilutions.
Pharmacologic Actions
Polyhexanide and chlorhexidine are taken up by amebic cysts and trophozoites.53 Cysts are more resistant than trophozoites.54 The effects depend on the concentration and duration of biocide.55
Biguanide disinfectants interrupt microbial DNA function by complexing with intracellular phosphated molecules such as adenosine triphosphate and nucleic acids. The cationic biguanides also adsorb to phospholipids of the trophozoite's plasma membrane, leading to cytoplasmic leakage. Biguanides may alter the permeability of amebic cysts in a similar way, resulting in shrinkage of intracystic amebae with separation of the plasma membrane from the endocystic wall.56 Biguanides also deform cytoplasmic organelles and cause nuclear chromatin to aggregate. By affecting the integrity of the cyst's ostiole, biocides might enhance the penetration of other antiparasitic agents. Whether resistance can be induced experimentally or by selective environmental exposure has not been determined. Several methods are available to assess the lethal effects of biguanides in vitro.57
Dosage and Pharmacokinetics
A 0.02% concentration of polyhexanide or chlorhexidine is typically used, at a frequency of every 1 to 2 hours during initial treatment. Corneal penetration kinetics have not been determined. Biguanides can bind to plastic and glass and to chloride anions in saline, but the clinical importance of these interactions has not been determined.
Adverse Effects
Stinging and burning may occur immediately after instillation. Conjunctival injection and punctate corneal epithelial erosions can occur. Dilute preparations less than 0.05% are safe for ocular application. Minimal toxicity of epithelial cells occurs with low concentrations of polyhexanide58 or chlorhexidine.59,60 High concentrations of biguanides cause toxic keratoconjunctivitis. Inappropriate use of 4% chlorhexidine in a detergent base can produce corneal inflammation and edema.61,62
Clinical Experience
One or the other of these two agents (polyhexanide and chlorhexidine) can be used in the initial treatment of Acanthamoeba keratitis. While some patients may respond to a single agent,62a others fail single-agent therapy.63 A biguanide is usually used in combination with other topical drugs such as propamidine or hexamidine.64,65 The addition of a topical biguanide to the regimen in patients unresponsive to other agents has led to clinical improvement.66 In vitro susceptibility, however, may not predict the clinical effectiveness of biguanide therapy.67 Insufficient information is available to judge the relative efficacy and safety of polyhexanide and chlorhexidine. The use of both agents together has not been assessed, and the role of other biocides (e.g. povidone-iodine) is unclear.
IMIDAZOLES AND TRIAZOLES
Historical Development
Imidazoles were developed in the 1960s at Farbenfabriken Bayer as synthetic organic chemicals. The antiprotozoal drug thiabendazole was found to have in vitro antifungal activity in 1965, and this led to the development of other imidazoles. The first compound studied for human fungal infections was clotrimazole.68 In vitro studies subsequently showed that miconazole and clotrimazole were effective agents against Acanthamoeba strains.69,70 Clotrimazole and ketoconazole were used in some of the first reported cases of Acanthamoeba keratitis.19,71 Imidazoles such as ketoconazole, miconazole, and clotrimazole have been used in the oral and topical treatment of Acanthamoeba keratitis. Triazoles such as itraconazole, fluconazole, and secnidazole have been recently introduced.
Chemistry and Preparations
Several imidazole and triazole compounds are commercially available (Fig. 4). Miconazole was available as a 20-mg/ml acidic solution, containing parabens, for intravenous infusion. This preparation has been dispensed, undiluted, in eyedropper bottles for topical use. Clotrimazole is available in dermatologic (Lotrimin and others) and vaginal (Mycelex and others) preparations, but these are irritating to the ocular surface. Topical clotrimazole can be prepared by obtaining the powder, ensuring sterility, and making a suspension in artificial tears.72 Clotrimazole 1% has been prepared from the micronized powder used for fungal sensitivity testing (Schering Corporation, Kenilworth, NJ). The suspension should be shaken before each use and may need to be reformulated every few weeks.
|
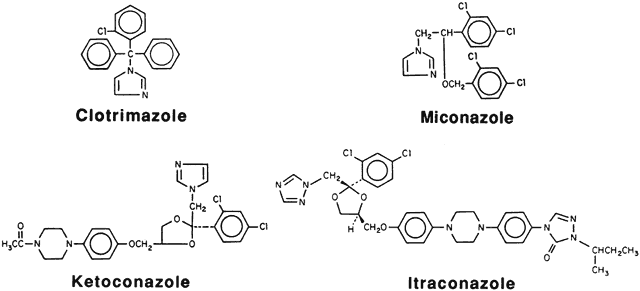
Several azole compounds are marketed for oral use. Ketoconazole is a weak dibasic imidazole piperazine compound that is practically insoluble in water at pH above 3. The drug is available as 200-mg scored tablets. Ophthalmic application of the 2% dermatologic cream can lead to a marked burning sensation. A topical preparation can be made by grinding the tablet to a fine powder and suspending it in buffered solution to yield a 2% concentration. Oral itraconazole is available as 100-mg capsules. The role of topical itraconazole or fluconazole in Acanthamoeba keratitis has not been studied.
Pharmacologic Actions
Imidazoles and triazoles exert an antifungal effect by disrupting the permeability of the cell membrane. Their mechanism of action on amebic trophozoites and cysts has not been adequately studied but presumably results in leakage of cytoplasmic molecules by interacting with the hydrophobic portion of the phospholipid cell membrane. Organisms that encyst become relatively more resistant to antiamebic agents.
Dosage and Pharmacokinetics
Oral absorption depends on gastric acidity, and bioavailability is reduced by H2-blockers such as ranitidine (Zantac) and famotidine (Pepcid) and by antacids. Its absorption in patients with achlorhydria can be enhanced by taking the drug with an acidic (pH 2.5) cola. The usual adult dose of ketoconazole is 400 mg daily, given as one or two doses. The oral drug is metabolized by the liver. By competing for hepatic microsomal enzymes, ketoconazole can increase the toxicity of other drugs, such as the nephrotoxicity of cyclosporine and the anticoagulant effect of warfarin. Oral ketoconazole results in a corneal level of approximately 50 μg/g that persists for at least 1 day.73
The usual oral dose of itraconazole is 200 mg daily, taken as one or two doses. Absorption of itraconazole is greater when taken with food. Hepatic metabolism of the triazoles is slower than that of the imidazoles.
Topical ophthalmic preparations of imidazoles are not commercially available but can be made with an appropriate vehicle. The lipid solubility of many azole compounds limits their ophthalmic use, although oils and cyclodextrins have been used to prepare these compounds.
Adverse Effects
Nausea is the most common side effect of ketoconazole and itraconazole after oral dosing and can be reduced by taking the tablet with meals. Itching and skin rash can occur. Inhibition of steroid biosynthesis can result in menstrual irregularities, male gynecomastia, and fluid retention. Drug-induced hepatitis is rare, but patients with anorexia, malaise, nausea, or abdominal pain should probably have liver function testing. Ketoconazole should be avoided in pregnant or nursing women. Compared with ketoconazole, the systemic triazoles have less effect on human sterol metabolism. Oral antihistamines such as astemizole (Hismanal) should be avoided because of the risk of serious cardiac dysrhythmia. The dosage of digoxin or oral hypoglycemic agents needs to be monitored during coadministration with oral ketoconazole or itraconazole. Cholesterol-lowering statins (e.g., lovastatin) may need to be discontinued during itraconazole therapy to avoid rhabdomyolysis.
The topical dermatologic and vaginal preparations of clotrimazole, miconazole, and related compounds may be too irritating for ophthalmic use. A 1% preparation of miconazole, clotrimazole, or ketoconazole produces minimal epithelial toxicity.74,75 Topical ketoconazole can precipitate on the eyelid margins and within a corneal epithelial defect.
Clinical Experience
Topical miconazole76–78 and topical clotrimazole79 have been successfully used in combination therapy. Adverse effects on the ocular surface limit their use in some patients. The benefit of oral ketoconazole or itraconazole in medical treatment is difficult to assess. Some ophthalmologists use an oral agent, taken with meals, as a supplement during the first few weeks of concomitant topical treatment.80