1. Hessen MT, Kaye D: Principles of selection and use of antibacterial agents in vitro activity
and pharmacology. Infect Dis Clin N Am 14:265–79, 2000. 2. Van Vlem B, Vanholder R, De Paepe P et al: Immunomodulating effects of antibiotics: literature review. Infection 24:275–291, 1996. 3. Labro MT: The prohost effect of antimicrobial agents as a predictor of clinical outcome. J Chemother 9:100–108, 1997. 4. Paladino JA: Pharmacoeconomics of antimicrobial therapy. Ame J Health Syst Pharm 56:S25–S28, 1999. 5. Wisniewski SR, Hammer ME, Grizzard WS et al: An investigation of the hospital charges related to the treatment of endophthalmitis
in the Endophthalmitis Vitrectomy Study. Ophthalmology 104:757–758, 1997. 6. World Health Organization (WHO): Antibiotic resistance a growing
threat, WHO reports. Improper drug use to blame [press release]. June 12, 2000. Available at: http://www.who.org 7. Centers for Disease Control and Prevention (CDC): Antibiotic
resistance. Available at: http://www.cdc.gov 8. Worakul N, Robinson J: Ocular pharmacokinetics-pharmacodynamics, Eur J Pharmaceut Biopharmaceut 44:71–83, 1997. 9. Meredith TA: Antibiotics and Antifungals. In Zimmerman TJ (ed): Textbook of Ocular Pharmacology, pp 363–385. Philadelphia: Lippincott-Raven1997. 10. Boothe DM: Small Animal Clinical Pharmacology and Therapeutics, pp 131–136. Philadelphia, WB Saunders, 2001. 11. Amsterdam D: Susceptibility testing of antimicrobials in liquid media. In Lorian V (ed): Antibiotics in Laboratory Medicine, pp 52–111. 4th ed. Baltimore, Williams & Wilkins, 1996. 12. Levison ME, Bush LM: Pharmacodynamics of antimicrobial agents. Bactericidal and postantibiotic effects. Infect Dis Clin North Am 3:415–421, 1989. 13. Ellner PD, Neu HC.: The Inhibitory Quotient A method for interpreting minimum inhibitory concentration
data:JAMA 246:1575–1578, 1981. 14. Estes L: Review of pharmacokinetics and pharmacodynamics of antimicrobial agents. Mayo Clinl Proc 73:1114–1122, 1998. 15. Forrest A, Nix DE, Ballow CH et al: Pharmacodynamics of intravenous ciprofloxacin in seriously ill patients. Antimicrob Agents Chemother 37:1073–1081, 1993. 16. Kays MB, Conklin M: Comparative in vitro activity and pharmacodynamics of five fluoroquinolones
against clinical isolates of Streptococcus pneumoniae. Pharmacotherapy 20:1310–1317, 2000. 17. Firsov AA, Vostrov SN, Shevchenko AA et al: A new approach to in vitro comparisons of antibiotics in dynamic models: Equivalent
area under the curve/MIC breakpoints and equiefficient doses
of trovafloxacin and ciprofloxacin against bacteria of similar susceptibilities,Antimicrob Agents Chemother; 42:2841–2847, 1998. 18. Craig WA, Gudmundsson S: Postantibiotic Effect. In Lorian V (ed): Antibiotics in Laboratory Medicine, pp 296–329. 4th ed. Baltimore, Williams & Wilkins, 1996. 19. Morlet N, Graham GG, Gatus B: Pharmacokinetics of ciprofloxacin in the human eye: A clinical study and
population pharmacokinetic analysis. Antimicrob Agents Chemother 44:1674–1679, 2000. 20. Bertino J, Drusano G, Kashuba ADM et al: Inability of a commercially available antibiotic utilization information
consultation program (AUIC) to predict outcome (OUT) or
time to event (TTE) in gram negative nosocomial pneumonia (GNNP). Proceedings of the 39th Interscience Conference on Antimicrobial Agents
and Chemotherapy of the American Society for Microbiology, San Francisco, CA, September 1999: Abstract # 1007, p A247. 21. Schoenwald RD: Ocular pharmacokinetics. In Zimmerman TJ et al (eds): Textbook of Ocular Pharmacology, pp 119–138. Philadelphia, Lippincott-Raven, 1997. 22. Seal DV, Bron AJ, Hay J: Antimicrobial pharmacology for the eye. In Ocular Infection, Investigation and Treatment in Practice, pp 12–13. London, Martin Dunitz, Ltd, 1998. 23. Davies N: Biopharmaceutical considerations in topical ocular drug delivery. Clin Expl Pharmacol Physiol 27:558–562, 2000. 24. Leeming JP: Treatment of ocular infections with topical antibacterials. Clin Pharmacokinet 37:351–360, 1999. 25. Mishima S, Gassett A, Klyce SD Jr et al: Determination of the tear volume and tear flow. Invest Ophthal Vis Sci 5:264, 1966. 26. Schoenwald RD: Chemical delivery systems with enhanced pharmacokinetic properties. In Mitra AK (ed): Ophthalmic Drug Delivery Systems, pp 307–330. New York, Marcel Dekker, 1993. 27. Sieg JW, Robinson JR: Mechanistic studies on transcorneal penetration permeation of pilocarpine. J Pharm Sci 65:1816–1822, 1976. 28. Mindel JS, Smith H, Jacobs M et al: Drug reservoirs in topical therapy. Invest Ophthal Vis Sci 25:346–350, 1984. 29. Doan MG, Jensen AD, Dohlman CH: Penetration routes of topically applied eye medications. Am J Ophthalmol 85:383–386, 1978. 30. Ahmed I, Patton TF: Importance of the noncorneal absorption route in topical ophthalmic drug
delivery. Invest Ophthalmol Vis Sci 26:584–587, 1985. 31. Edelhauser HF, Maren TH: Permeability of human cornea and sclera to sulfonamide carbonic anhydrase
inhibitors. Arch Ophthalmol 106:1110–1111, 1988. 32. Chien DS, Honsy JJ, Gluchowski C et al: Corneal and conjunctival/scleral penetration of p-aminoclonidine, AGN 190342, and clonidine in rabbit eyes. Curr Eye Res 9:1051–1059, 1990. 33. Weng W, Sasaki H, Chien DS et al: Lipophilicity influence on conjunctival drug penetration in the pigmented
rabbit: A comparison with corneal penetration. Curr Eye Res 10:571–579, 1991. 34. Lee DY, Schoenwald RD, Barfknecht CF: Biopharmaceutical explanation for the topical activity of 6-hydroxyethoxy-2-benzothiazole-sulfonamide in the rabbit
eye. J Ocul Pharmacol 8:247–265, 1992. 35. Pech B, Chetoni P, Saettone MF et al: Preliminary evaluation of a series of amphiphilic timolol prodrugs: Possible
evidence for transscleral absorption. J Ocul Pharmacol 9:141–150, 1993. 36. Worakul N, Robinson J: Ocular pharmacokinetics-pharmacodynamics. Eur J Pharmaceut Biopharmaceut 44:71–83, 1997. 37. Patel G, Chuang A, Kiang E, et al: Epithelial healing rates with topical ciprofloxacin, ofloxacin, and ofloxacin
with artificial tears after photorefractive keratectomy. J Cataract Refract Surg 26:690–694, 2000. 38. Maurice DM: The tonicity of an eyedrop and its dilution by tears. Exp Eye Res 11:30, 1971. 39. Swan KD, White NG: Corneal permeability: I. Factors affecting penetration of drugs into the
cornea. Am J Ophthalmol 25: 1043, 1942. 40. Benson H: Permeability of the cornea to topically applied drugs. Arch Ophthalmol 91:313, 1974. 41. McDermott ML, Tran TD, Cowden JW et al: Corneal stromal penetration of topical ciprofloxacin in humans. Ophthalmology 100:197–200, 1993. 42. O'Brien TP, Sawuch MR, Dick JD et al: Topical ciprofloxacin treatment of Pseudomonas keratitis in rabbits. Ophthalmology 106:1444–1446, 1988. 43. Barza M, Lynch E, Baum JL: Pharmacokinetics of newer cephalosporins after subconjunctival and intravitreal
injection. Arch Ophthalmol 111:121–125, 1993. 44. Shockley RK, Jay WM, Fishman PH, et al: Effect of inoculum size on the induction of endophthalmitis in aphakic
rabbit eyes. Acta Ophthalmol (Copenhag) 63:35–38, 1985. 45. Maylath FR, Leopold PH: Study of experimental intraocular infection. I. The recoverability of organisms inoculated into ocular tissue and fluids. II. The
influence of antibiotics and cortisone, alone and combined, on
intraocular growth of these organisms. Am J Ophthalmol 40:86–101, 1955. 46. John T, Sims M, Hoffmann C: Intraocular bacterial contamination during sutureless, small incision single-port
phacoemulsification. J Cataract Refract Surg 26:1786–1791, 2000. 47. Egger SF, Huber-Spiezy V, Scholda C et al: Bacterial contamination during extracapsular cataract extraction; prospective
study on 200 consecutive patients. Ophthalmologica 208:77–81, 1994. 48. Barza M: Antimicrobial barriers for antimicrobial agents. Eur J Clin Microbiol Infect Dis 12:S31–S35, 1993. 49. Barza M: Treatment of eye infections. In Hooper DC, Wolfson JS (eds): Quinolone Antimicrobial Agents, pp 423–424. 2nd ed. Washington, DC, American Society for Microbiology, 1993. 50. Donnenfeld ED, Perry HD, Snyder RW et al: Intracorneal, Aqueous humor, and vitreous humor penetration of topical
and oral ofloxacin. Arch Ophthalmol 115:173–176, 1997. 51. Fiscella RG, Nguyen TKP, Cwik MJ et al: Aqueous and vitreous penetration of levofloxacin after oral administration. Ophthalmology 106:2286–2290, 1999. 52. American Society of Health-System Pharmacists: Anti-infective
agents. In: McEvoy GK, Litvak K, Welsh OH Jr et al (eds): American Hospital Formulary Service (AHFS) Drug Information. Bethesda, MD,1: American Society of Health-System Pharmacists, 2000. 53. Bush LM, Johnson CC: Ureidopenicillins and beta-lactam/beta-lactamase inhibitor
combinations. Infect Dis Clin N Am 14:409–433, 2000. 54. Asbel LE, Levison ME: Cephalosporins, carbapenems, and monobactams. Infect Dis Clin N Am 14:435–447, 2000. 55. Patalano SM, Hyndiuk RA: Aminoglycosides in ophthalmology. In ZimmermanTJ et al: Textbook of Ocular Pharmacology, pp 531–535. Philadelphia, Lippincott-Raven, 1997. 56. Chan-Tack KM: Changing antibiotic sensitivity patterns at a university hospital, 1992 through 1999. South Med J 94:619–620, 2001. 57. Milazzo G, Papa V, Carstocea B et al: Topical netilmicin compared with tobramycin in the treatment of external
ocular infection. Int J Clin Pharmacol Ther 37:243–248, 1999. 58. Daikos GL, Jackson GG, Lolans VT et al: Adaptive resistance to aminoglycoside antibiotics from first-exposure
down-regulation. J Infect Dis 162:414–420, 1990. 59. Widdowson CA, Klugman KP: Molecular mechanisms of resistance to commonly used non-beta lactam
drugs in Streptococcus pneumoniae. Semin Respir Infect 14:255–268, 1999. 60. Crumplin GC: The Mechanism of Action of Quinolones: Quinolones—Their Future in
Clinical Practice, pp 1–14. London, Royal Society of Medicine Services; 1986. 61. Gwon A: Ofloxacin vs tobramycin for the treatment of external ocular infection. Arch Ophthalmol 110:1234–1237, 1992. 62. Drugeon HB, Juvin ME, Bryskier A: Relative potential for selection of fluoroquinolone-resistant Streptococcus pneumoniae strains by levofloxacin: Comparison with ciprofloxacin, sparfloxacin, and
ofloxacin. J Antimicrobiol Chemother 43(SupplC):55–59, 1999. 63. Goldstein MH, Kowalski RP, Gordon YJ: Emerging fluoroquinolone resistance bacterial keratitis: a 5-year
review. Opththalmology 106:1313–1318, 1999. 64. Schentag JJ, Nix DE, Forrest A: Pharmacodynamics of Fluoroquinolones. In Hooper DC, Wolfson JS (eds): Quinolone Antimicrobial Agents, pp 259–271. 2nd ed. Washington, DC, American Society for Microbiology, 1993. 65. Antibiotic Update—Current Treatment Modalities. In: Ocular Surgery News, January 1, 2000 supplement, p. 12. 66. Prober GC: Bacterial resistance and the dilemma of antibiotic usage. West J Med. 1997; 166:337–338. 67. Ogawa GSH, Hyndiuk RA: The fluoroquinolones: New antibiotics in ophthalmology. Int Ophthalmol Clin 33:59–68. 68. Martínez-Martínez L, Pascual A, Jacoby GA: Quinolone resistance from a transferable plasmid. Lancet 351:797–799, 1998. 69. Robert PA, Adenis JP: Comparative review of topical ophthalmic antibacterial preparations. Drugs 61:175–185, 2001. 70. Zuckerman JM: The newer macrolides: azithromycin and clarithromycin. Infect Disease ClinN Am 14:449–462. 71. Setti EL, Micetich RG: New trends in antimicrobial development. Curr Med Chem 5:101–113, 1998. 72. Centers for Disease Control and Prevention: Update: Staphylococcus aureus with reduced susceptibility to vancomycin—United States, 1997. MMWR. Morb Mortal Wkly Rep 46:813–815, 1997. 73. Centers for Disease Control and Prevention: Update: Staphylococcus aureus with reduced susceptibility to vancomycin—Japan, 1997. MMWR. Morb Mortal Wkly Rep 46:765, 1997. 74. Lundstrom TS, Sobel JD: Antibiotics for gram-positive bacterial infections: Vancomycin, teicoplanin, quinupritin/dalfopristin, and linezolid. Infect Dis Clin N Am 14:463–474, 2000. 75. Epstein ME, Amodio-Groton M, Sadick NS: Antimicrobial agents for the dermatologist. II. Macrolides, trimethoprim-sulfamethoxazole
and clindamycin. JAm Acad Dermatol 37:365–381, 1997. 76. Garcia F, Blanco J, Carretero P et al: Anaphylactic reactions to topical rifamycin. Allergy 54:527–528, 1999. 77. Breithaupt H: The new antibiotics. Nat Biotechnol 17:1165–1169, 1999. 78. Mitsui Y, Ooishi M, Sasaki K et al: AQCmax as a pharmacokinetic parameter of ophthalmic solution. Jpn J Eye 12:783–786, 1995. 79. Bucci FA: An in vivo comparison of the ocular absorption of levofloxacin versus ciprofloxacin
prior to phacoemulsification. The Association for Research in Vision and Ophthalmology Annual Meeting. Poster B589, Presentation 1579, May, 2002. 80. Bucci FA, O'Brien TP, Evans RE et al: A prospective comparison of four methods of pre-phacoemulsification
antibiotic treatment with ofloxacin and ciprofloxacin [abstract]. Invest Ophthalmol Vis Sci 41:5768, 2000. 81. Hoogkamp-Korsranje JAA, Roelofs-Willemse J: Comparative in vitro activity of moxifloxacin against Gram-positive clinical isolates. J Antimicrob Chemother 45:31–39, 2000. 82. Fung-Tomc J, Gradelski E, Huczko E et al: Activity of gatifloxacin against strains resistant to ofloxacin and ciprofloxacin
and its ability to select for less susceptible bacterial variants. Int J Antimicrob Agents 18:77–80, 2001. 83. Tsurumaki Y, Manda H, Takei M et al: In vitro antimicrobial activity of gatifloxacin against 873 clinical isolates
from respiratory tract, urinary tract and surgical infections during 1997–1998 in
Japan. Journal of American Chemotherapy 45:685–689, 2000. 84. Jones ME, Staples AM, Critchley I et al: Benchmarking the in vitro activity of moxifloxacin against recent isolates of Streptococcus pneumoniae, Moraxella catarrhalis, and Haemophilus influenzae. A European multi-center study. Diagn Microbiol Infect Di 37:203–211, 2000. 85. Huczko E, Conetta B, Bonner D et al: Susceptibility of bacterial isolates to gatifloxacin and ciprofloxacin
from clinical trials 1997–1998. Int J Antimicrob Agents 16:401–405, 2000. 86. Jones ME, Angel M, Staples AM et al: Benchmarking the in vitro activity of moxifloxacin and comparator agents against recent respiratory
isolates from 377 medical centers throughout the United States. Antimicrob Agents Chemother 44:2645–2652, 2000. 87. Hoban DJ, Bouchilon SK, Karlowsky JA et al: A comparative in vitro surveillance study of gemifloxacin activities against 2632 recent Streptococcus pneumoniae isolates from across Europe, North America, and South America. The Gemifloxacin
Surveillance Study Research Group. Antimicrob Agents and Chemother 44:3008–11, 2000. 88. Ieven M, Goossens W, De Wit S et al: In vitro activity of gemifloxacin compared with other antimicrobial agents
against recent clinical isolates of streptococci. J Antimicrob Chemother 45(Suppl1):51–53, 2000. 89. Lemmen S, AlpLahham A, Utticken R et al: In vitro activity of gemifloxacin against Streptococcus pneumoniae isolates in Germany. Chemotherapy 45:104–109, 2000. 90. Biedenbach DJ, Jones RN, Pfaller MA et al: Activity of BMS284756 against 2681 recent clinical isolates of Haemophilus influenzae and Moraxella catarrhalis: report from the SENTRY Antimicrobial Surveillance Program (2002) in
Europe, Canada, and the United States. Diagn Microbiol Infect Dis 39:245–250, 2000. 91. Lewis MT, Jones RN: Activity of macrolides, lincosamines, streptgramins and fluoroquinolones
against Streptococcus pneumoniae and Enterococci isolated from the Western Hemisphere: Example of international surveillance (SENTRY
Antimicrobial Surveillance Program) in the development
of new drugs. Braz J Infect Dis 4:15–21, 2000. 92. Dubois J, St.-Pierre C: In vitro activity of ABT-773 versus macrolides and quinolones against resistant
respiratory tract pathogens. Diagn Microbiol Infect Dis 40:35–40, 2001. 93. Betriu C, Redondo M, Palau ML et al: Comparative in vitro activities of linezolid, quinupristin-dalfopristin, moxifloxacin, and
trovafloxacin against erythromycin-susceptible and -resistant
streptococci. Antimicrob Agents Chemother 44:1838–1841, 2000. 94. Doern GV, Heilmann KP, Huynh HK et al: Antimicrobial resistance among clinical isolates of Streptococcus pneumoniae in the United States during 1999–2000, including a comparison of
resistance rates since 1994–1995. Antimicrob Agents Chemother 45:1721–1729, 2001. 95. Cercenado E, Garcia-Garrot F, Bouza E: In vitro activity of linezolid against multiple resistant Gram-positive
clinical isolates. J Antimicrob Chemother 47:77–81, 2001. 96. Hoban DJ, Bouchillon SK, Johnson JL et al: Comparative in vitro activity of gemifloxacin, ciprofloxacin, levofloxacin and ofloxacin in
a North American surveillance study. Diagn Microbiol Infect Dis 40:51–57, 2001. 97. Bauernfeind A: Comparison of the antibacterial activities of the quinolones BAY 12-8039, gatifloxacin (AM 1155), trovafloxacin, clinafloxacin, levofloxacin
and ciprofloxacin. J Antimicrob Chemother 40:639–651, 1997. 98. Souli M, Wennersten CB, Eliopoulos GM: In vitro activity of BAY 12-8039, a new fluoroquinolone, against species
representative of respiratory tract pathogens. Int J Antimicrob Agents 10:23–30, 1998. 99. Fass RJ: In vitro activity of BAY 12-8039, a new 8-methoxyquinolone. Antimicrob Agents Chemother 41:1818–1824, 1997. 100. Woodcock JM, Andrews JM, Boswell FJ et al: In vitro activity of BAY 12-8039, a new fluoroquinolone. Antimicrob Agents Chemother 41:101–106, 1997. 101. Wakabayashi E, Mitsuhashi S: In vitro antibacterial activity of AM-1155, a novel 6-fluoro-8-methoxy
quinolone. Antimicrob Agents Chemother 38:594–601, 1994. | 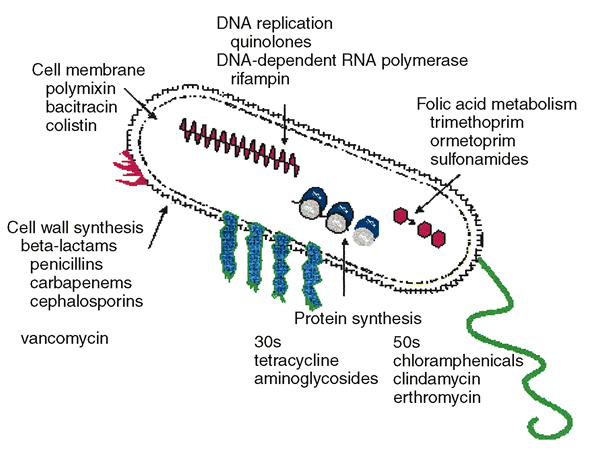
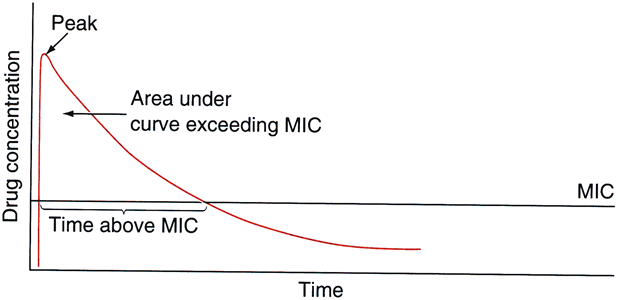
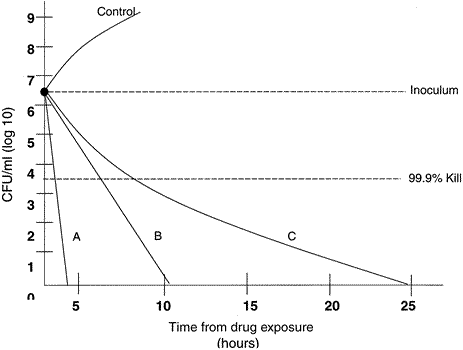
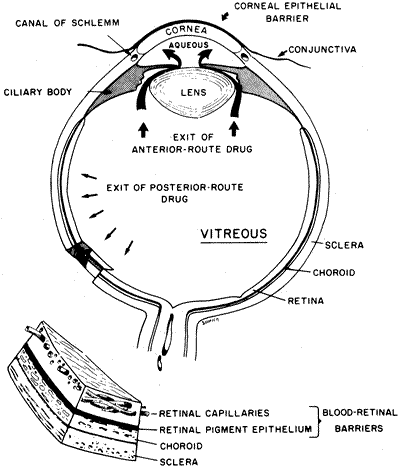
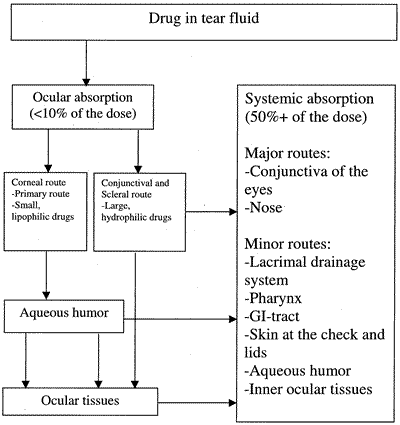
 32)
32) 0.03)
0.03)