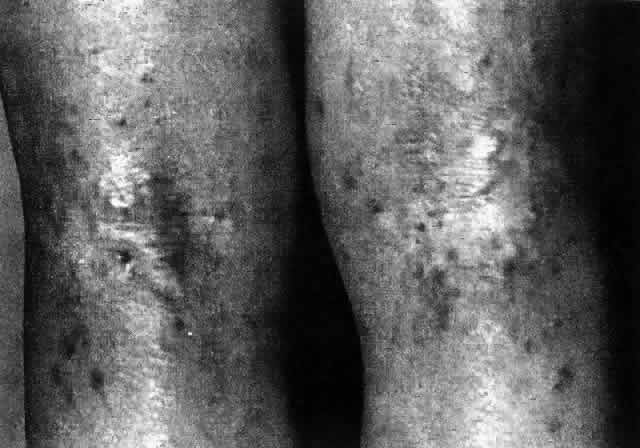
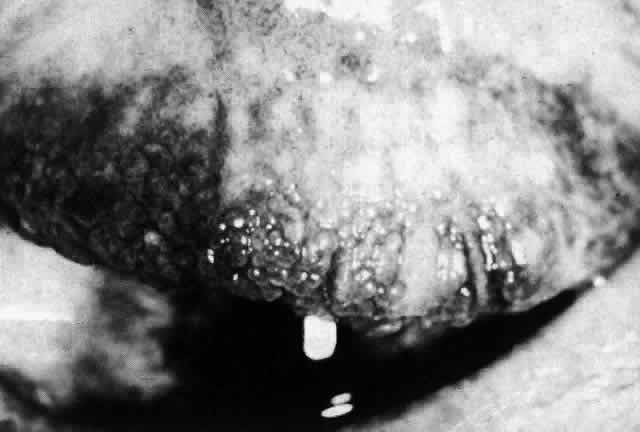
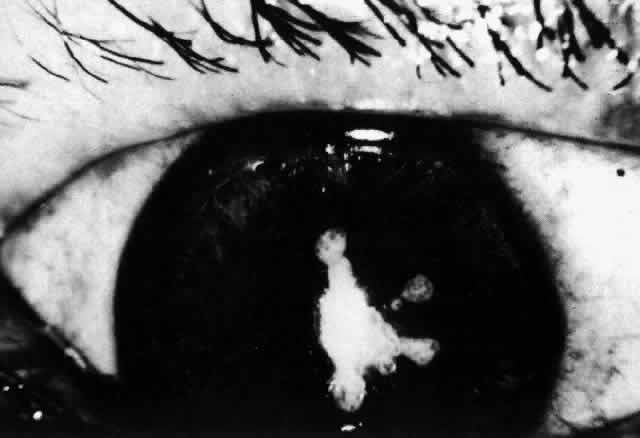
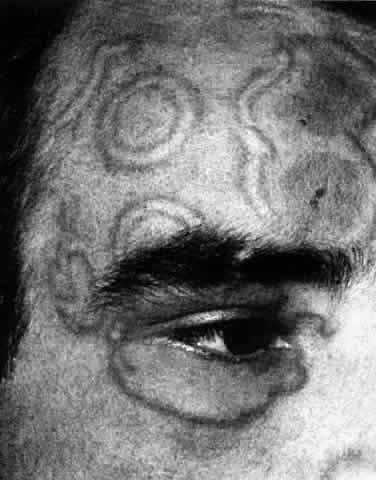
1. Dogru M, Nakagawa N, Tetsumoto K et al: Ocular surface disease in atopic dermatitis. Jpn J Ophthalmol 43:53, 1999 1a. Winkelman RK: Nonallergic factors in atopic dermatitis.J Allergy 37:29, 1966 2. Lobitz WC Jr, Campbell DJ: Physiologic studies in atopic dermatitis (disseminated neurodermatitis): I. The
local cutaneous response to intradermally injected acetylcholine
and epinephrine. Arch Dermatol 67:575, 1953 3. Blomgren SE: Erythema nodosum. Semin Arthritis Rheum 4:1, 1974 4. Solomon LM, Nadler NJ: Radioautography of nor-adrenaline-14 in atopic dermatitis. Can Med Assoc J 96:1147, 1967 5. Stone SP, Muller SA, Gleich GJ: IgE levels in atopic dermatitis. Arch Dermatol 108:806, 1973 6. Johannson SGO, Juhlin L: Immunoglobulin E in healed atopic dermatitis and after treatment with corticosteroids
and azathioprine. Br J Dermatol 82:10, 1970 7. McGready SJ, Buckley RH: Depression of cell-mediated immunity in atopic eczema. J Allergy Clin Immunol 56:393, 1975 8. Rachelefsky GS, Opelz G, Mickey MR et al: Defective T cell function in atopic dermatitis. J Allergy Clin Immunol 57:569, 1976 9. Lobitz WC, Honeyman JF, Winkler NW: Suppressed cell mediated immunity in two adults with atopic dermatitis. Br J Dermatol 86:317, 1972 10. Wenner HA: Complications of infantile eczema caused by the virus of herpes simplex. Am J Dis Child 67:247, 1944 11. Brown MA, Hanifin JM: Atopic dermatitis. Curr Opin Immunol 2:531, 1990 11a. Hingorani M, Calder V, Jolly G et al:Eosinophil surface antigen expression and cytokine production vary in different
ocular allergic diseases.J Allergy Clin Immunol 10:821, 1998 11b. Morgan SJ, Williams JH, Walls AF et al:Mast cell hyperplasia in atopic keratoconjunctivitis: An immunohistological
study.Eye 5:729,1991 11c. Lee SJ, Rice BA, Foster CS: HLA class II expression in normal and inflamed conjunctiva.Reg Immunol 3:177,1990-91 11d. Bielory L: Contact dermatitis of the eye.Immunol Allergy Clin North Am 17:131, 1997 12. Dahl MV: Chronic, irritant contact dermatitis: Mechanisms, variables, and differentiation
from other forms of contact dermatitis. Adv Dermatol 3:261, 1988 13. Landsteiner K, Chase MW: Experiments on transfer of cutaneous sensitivity to simple compounds. Proc Soc Exp Biol Med 71:516, 1942 14. Dvorak HF, Dvorak AM: Basophilic leukocytes: Structure, function and role in disease. Clin Haematol 4:651, 1975 15. Dvorak HF, Dvorak AM, Simpson BA et al: Cutaneous basophil hypersensitivity: II. A light and electron microscopic
study. J Exp Med 132:558, 1970 15a. Cooper KD: Urticaria and angioedema: Diagnosis and evaluation. J Am Acad Dermatol 25:166, 1991 15b. Greaves M, Lawlor F, Angioedema: Manifestatons and management. Medicine 43:131, 1964 16. Beall GN: Urticaria: A review of laboratory and clinical observations. Medicine 43:131, 1964 17. Greaves MW, Plummer VM, McLaughlin P et al: Serum and cell bound IgE in
chronic urticaria Clin Allergy 4:265, 1974 18. Buckley RH, Dees SC: Serum immunoglobulins: III. Abnormalities associated with chronic urticaria
in children. J Allergy 40:294, 1967 19. Osler W: Hereditary angioneurotic edema. Am J Med Sci 95:362, 1888 20. Austen KF, Sheffer AL: Detection of hereditary angioneurotic edema by demonstration of a reduction
in the second component of human complement. N Engl J Med 272:649, 1965 21. Donaldson V, Evans RR: A biochemical abnormality in hereditary angioneurotic edema. Am J Med 35:37, 1963 22. Ruddy S, Gigli I, Austen KF: The complement system of man. N Engl J Med 287:489, 545, 592, 632, 1972 23. Frank MM, Gelfand JA, Atkinson JP: Hereditary angioedema: The clinical syndrome and its management. Ann Intern Med 84:580, 1976 24. Sim TC, Grant JA: Hereditary angioedema: Its diagnostic and management perspectives. Am J Med 88:656, 1990 25. Kaldeck R: Ocular psoriasis. Arch Dermatol 68:44, 1953 26. Russell TF, Schultes LM, Kuban DJ: Histocompatibility (HL-A) antigens associated with psoriasis. N Engl J Med 287:738, 1972 27. White SH, Newcomer V, Mickey M et al: Disturbance of HL-A antigen frequency in psoriasis. N Engl J Med 287:740, 1972 28. Jordan RE, Beutner EH, Witebsky E: Basement zone antibodies in bullous pemphigoid. JAMA 200:751, 1971 29. Jordan RE, Schoeter AK, Rogers RS III et al:Classical and alternate pathway activation of complement in pemphigus vulgaris
lesions. J Invest Dermatol 63:256, 1974 30. Epstein WL, Maibach HI: Immunologic competence of patients with psoriasis receiving cytotoxic therapy. Arch Dermatol 91:599, 1965 31. Levantine A, Brostoff J: Immunological responses of patients with psoriasis and the effect of treatment
with methotrexate. Br J Dermatol 93:659, 1975 32. Dolan PA, Flowers FP, Arujo OE et al: Toxic epidermal necrolysis. J Emerg Med 7:65, 1989 33. Koblenzer PJ: Toxic epidermal necrolysis (TEN, Ritter's disease) and staphylococcal
scalded skin syndrome (SSSS): A description and review. Clin Pediatr Phila 15:724, 1976 34. Manganotti G, Silvestri U: Contributo allo studio della sindrome di Lyell. Arch Ital Dermatol 32:192, 1964 35. Husz S, Berko G, Schneider I et al: Immunoelectrophoretic changes in Lyell syndrome. Int J Dermatol 13:205, 1974 36. Stein J, Turk E: Beitrag zum Epidermolyse-Syndrome (Lyell) im Kindesalter. Dtsch Ges Wes 22:2125, 1963 37. Beutner EH, Lever WF, Witebsky E et al: Autoantibodies in pemphigus vulgaris. JAMA 192:682, 1965 38. Bean SF, Holubar K, Gillett RB: Pemphigus involving the eyes. Arch Dermatol 111:1484, 1975 39. Beutner EH, Jordan RE: Demonstration of skin antibodies in serum of pemphigus vulgaris patients
by indirect immunofluorescent staining. Proc Soc Exp Biol Med 117:505, 1964 40. Wood GW, Beutner EH, Chorzelski TP: Studies in immunodermatology: II. Production of pemphigus-like lesions
by intradermal injection of monkeys with Brazilian pemphigus foliaceous
sera. Int Arch Allergy 42:456, 1972 41. Sams WM, Jordan RE: Pemphigus antibodies: Their role in disease. J Invest Dermatol 56:474, 1971 42. Wichmann JE: Ibdeen zur Diagnostik: Beobachtenden Aertzen Mitgetheilet. Vol 1. Hanover, Germany, Helwing, 1794, p 89 43. Cooper W: Pemphigus of the conjunctiva. Ophthalmol Hosp Rep 1:155, 1858 44. Von Graefe A, cited in Taylor RF: Modern treatment of severe shrinkage
of the conjunctiva. Br J Ophthalmol 51:31, 1967 45. Thost A: Der chronische Schleimhautpemphigus der oberen Luftwege. Arch Otorhinolaryngol 25:459, 1911 46. Civatte A: Le diagnostic des dermatoses buleuses au laboratoire. Arch Belg Derm Syph 5:273, 1949 47. Lever WF: Pemphigus: A histopathologic study. Arch Dermatol 64:727, 1951 48. Peck SM, Osserman KE, Weiner LB et al: Studies in bullous diseases: Immunofluorescent serologic tests. N Engl J Med 279:951, 1968 49. Jablonska S: Immunopathology of bullous diseases. Ann Clin Res 2:7, 1970 50. Bean SF, Waisman M, Michel B et al: Cicatricial pemphigoid. Arch Dermatol 106:195, 1972 51. Furey N, West C, Andrews T et al: Immunofluorescent studies of ocular cicatricial pemphigoid. Am J Ophthalmol 80:825, 1975 52. Herron BE: Immunologic aspects of cicatricial pemphigoid. Am J Ophthalmol 79:271, 1975 53. Person J, Rogers RS: Bullous and cicatricial pemphigoid: Clinical, histopathologic and immunopathologic
correlations. Mayo Clin Proc 52:54, 1977 54. Waltman S, Yarian D: Circulating autoantibodies in ocular pemphigoid. Am J Ophthalmol 77:891, 1974 55. Bean SF: Cicatricial pemphigoid. Int J Dermatol 14:23, 1975 56. Cram DL, Griffith MR, Fukuyama K: Pemphigus-like antibodies in cicatricial pemphigoid. Arch Dermatol 109:235, 1974 56a. Elder MJ, Lightman S: The immunological features and pathphysiology of ocular cicatrical pemphigoid.Eye 8:196, 1994 56b. Bernauer W, Wright P, Dart JK et al:The conjunctiva in acute and chronic mucous membrane permphigoid: An immunohistochemical
analysis. Ophthalmology 100:339, 1993 56c. Heilinghaus A, Schaller J, Mauss S et al:Eosinophil granule proteins expressed in ocular cicatricial pemphigoid. Br J Ophthalmol 82:312,1998 57. Patten JT, Cavanagh HD, Allansmith MR: Induced ocular pseudopemphigoid. Am J Ophthalmol 82:272, 1976 58. Norn MS: Pemphigoid related to epinephrine treatment. Am J Ophthalmol 83:138, 1977 59. Glowa JR, Barrett JE: Drug history modifies the behavioral effects of pentobarbital. Science 220:333, 1983 60. Beutner EH, Chorzelski TB, Jordan RE: Autosensitization in Pemphigus and
Bullous Pemphigoid. Springfield, Illinois, Charles C Thomas, 1971, p 143 61. Provost TT, Tomasi TB: Immunopathology of bullous pemphigoid. Clin Exp Immunol 18:193, 1974 62. Ishizaka T, Sian CM, Ishizaka K: Complement fixation by aggregated IgE through alternate pathway. J Immunol 108:848, 1972 63. Arbesman CE, Wypych JI, Reisman RE: Serum IgE in human diseases. In Goodfriend
L, Sehon AH, Orange R (eds): Mechanisms in Allergy (Reagin Mediated
Hypersensitivity) Immunology (series). Vol 1. New York, Marcel Dekker, 1973, p 163 64. Sams WM, Jordan RE: Correlation of pemphigoid and pemphigus antibody titers with activity of
the disease. Br J Dermatol 84:7, 1971 65. Sams WM: Pemphigus and pemphigoid: Dermatologic example of type II immune reactivity. Ann Allergy 37:255, 1976 66. Chlorzelski TP, Beutner EH, Jablonska S et al: Immuno-fluorescence studies in the diagnosis of dermatitis herpetiformis
and its differentiation from bullous pemphigoid. J Invest Dermatol 56:373, 1971 67. Van Der Meer JB: Granular deposits of immunoglobulins in the skin of patients with dermatitis
herpetiformis: An immunofluorescent study. Br J Dermatol 84:493, 1969 68. Provost TT, Tomasi TB: Evidence for the activation of complement via the alternate pathway in
skin disease: II. Dermatitis herpetiformis. Clin Immunol Immunopathol 3:178, 1974 69. Gebhard RL, Falchuk ZM, Katz SI et al: Dermatitis herpetiformis: Immunologic concomitants of small intestinal
disease and relationship to histocompatibility antigen HL-A8. J Clin Invest 54:98, 1974 70. Jablonska S, Chorzelski TP, Beutner EH et al: Dermatitis herpetiformis and bullous pemphigoid. Arch Dermatol 112:45, 1976 71. Stevens AM, Johnson FC: A new eruptive fever associated with stomatitis and ophthalmia: A report
of two cases in children. Am J Dis Child 24:526, 1922 72. Foster CS, Fong LP, Azar D et al: Episodic conjunctional inflammation after Stevens-Johnson syndrome. Ophthalmology 95:4531, 1988 73. Shelley WB: Herpes simplex virus as a cause of erythema multiforme. JAMA 201:153, 1967 74. Whitmore PV: Skin and mucous membrane disorders. In Duane R (ed): Clinical
Ophthalmology. Vol 5. Philadelphia, JB Lippincott, 1980, pp 1–27 75. Mondino B: Cicatricial pemphigoid and erythema multiforme. Ophthalmology 97:939, 1990 76. Atkin N, Nitzberg S, Dorsey J: Lysis of intracerebral thromboembolism with fibrinolysin: Report of a case. Angiology 15:346, 1964 76a. Mondino BJ: Cicatricial pemphigoid and erythema multiforme. Ophthalmology 97:939, 1990 77. Kruger GG, Weston WL, Thorne EG et al: A phenomenon of macrophage aggregation activity in sera of patients with
exfoliative erythroderma, erythema multiforme, and erythema nodosum. J Invest Dermatol 60:282, 1973 |