|
PHYSIOCHEMICAL BARRIERS
Physiochemical barriers are significant first-line elements of a host's defense. The skin and mucous membranes limit access to the host by invading microorganisms. Enzymes, such as lysozyme, nonspecifically inhibit microbial growth. In the eye, the conjunctiva and the tear fluid layer provide the primary barrier against environmental aeroallergens, chemicals, and infectious agents. The tear fluid layer contains both specific and nonspecific immunologically active proteins, including lysozyme, histamine, tryptase, lactoferrin, ceruloplasmin, vitronectin, immunoglobulin A (IgA), immunoglobulin G (IgG), immunoglobulin M (IgM), and immunoglobulin E (IgE).4
Lymphocytes
Lymphocytes are the cells responsible for the specificity of immune recognition and for coordination of the immune response. They are derived from a lymphoid progenitor cell in the bone marrow and are divided into three classes: T cells (lymphocytes), B cells (lymphocytes), and natural killer (NK) cells. T cells and B cells are morphologically indistinguishable, small 8- to 10-μm-in-diameter lymphocytes with large nuclei. They are functionally distinct and are easily differentiated by the cell surface proteins they express: T cells with CD3, CD4, and CD8 and B cells with CD-19 surface markers.1
NK cells are large lymphocytes with many cytoplasmic granules, distinct cell surface markers (CD16), and the ability to lyse cells directly, especially tumor cells or normal cells infected by virus. NK cells lack immunoglobulin or TCRs for antigen recognition yet do not kill their targets at random. Their means for target cell recognition is not well understood. NK cells do participate in antibody-dependent cellular cytotoxicity (ADCC) on the basis of their expression of a low-affinity receptor for the Fc portion of IgG (CD16). In ADCC, a target cell coated with IgG can be lysed directly by an NK cell after binding of the Fc portion of IgG with its low-affinity receptor CD16. This not only provides a means of recognition for the target by the NK cell but also serves to activate it to release its granule contents such as perforin, an enzyme that perforates the wall of the target cell, and secrete cytokines to augment the inflammatory response.1
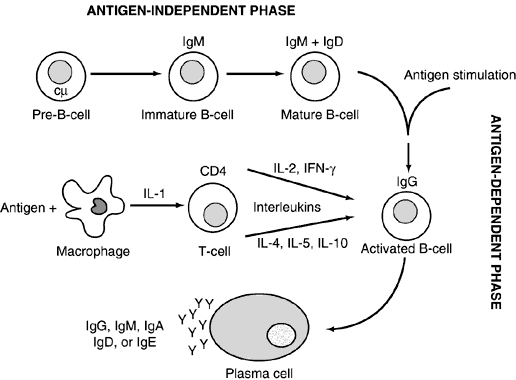
B cells develop from their precursors in the fetal liver and adult bone marrow. (They were first shown to mature in birds in the bursa of Fabricius, hence “B” cell.) They are primarily responsible for humoral immunity and are the exclusive producers of immunoglobulins, also known as antibodies, thus playing a vital role in the recognition and elimination of foreign antigen (further discussion on immunoglobulins below). Mature B cells can be divided into memory cells for the development of a rapid secondary response and plasma cells, which are totally committed to produce a single protein, an immunoglobulin. Plasma cells are terminally differentiated producers of large amounts of antibody (Fig. 2). B cells also interact closely with helper T cells through cell surface proteins such as CD40 and class II MHC and their complementary ligands.
|
T cells develop from their bone marrow precursors in the thymus, where somatic gene rearrangement gives rise to functional TCR complexes as well as distinctive cell surface proteins. T cells can be functionally subdivided into either helper or cytolytic (CTL) cells. These functionally different populations express distinct cell surface proteins: CD4 on helper cells and CD8 on CTLs, which serve as ligands for the MHC gene products on antigen-presenting cells.5
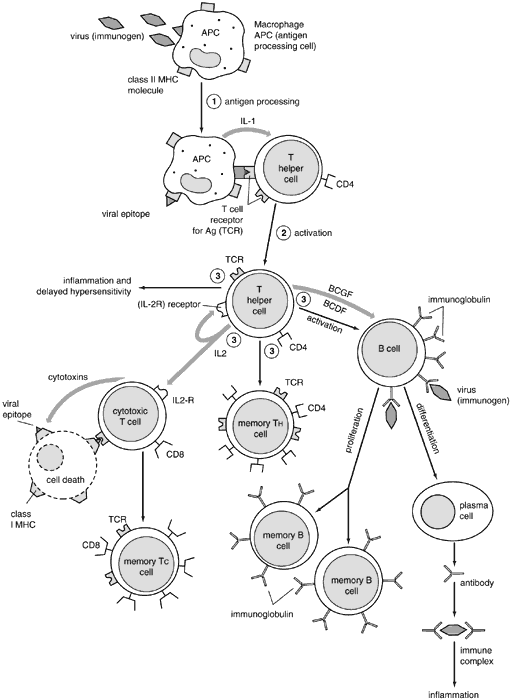
CD4+ cells play a vital role in B-cell growth and differentiation, including the production of antibodies.6 They also are crucial in macrophage activation and in upregulating or downregulating the immune response. CD4+ cells can be further subdivided into TH1 and TH2 lymphocyte populations based on their cytokine-producing properties. Cytokines are small proteins produced by cells in response to a variety of inducing stimuli. Cytokines are produced by their producer cells and then influence the behavior on target cells. Hormones are classical polypeptides that also fulfill this definition but are not by convention classified as cytokines since they are produced by specific endocrine organs (e.g., the thyroid gland), whereas a cytokine may be produced by more than one cell type in a number of different tissues. Cytokines similar to hormones act on the cells that produce them (autocrine), on cells in a distant organ (endocrine), or on cells in the immediate vicinity of their production (paracrine) fashion to regulate lymphocytes, antigen-presenting cells, other inflammatory cells, and immunologically active molecules involved in cellular communication. Multiple studies have shown that cytokines also are characterized by their ability to act on different cells (pleiotropism), produce different effects on the same target cell, share properties with other cytokines (redundancy), and influence the production of other cytokines.1 It also has been discovered that among their many functions, cytokines can affect cell growth and differentiation similarly to those of the growth factors. Given the enormous diversity of cells producing cytokines and the numerous effects (many of which still are unknown) exerted by these molecules, it has been difficult to develop a satisfactory classification (Table 1). Cytokines also may be classified by cell of origin, but it is now clear that certain cytokines are produced by many cells, making these criteria impractical for classification.
Antiviral Agents
Interferon <ga>, <gb>, <gg>
Colony-stimulating factors
CSF-1 or M-CSF
GM-CSF
G-CSF
IL-3
Stem cell factors
Erythropoietin
Thrombopoietin
Lymphoid growth factors
IL a,b
IL-2-15
Growth promoters
Bone morphogeneic protein
Ciliary neurotrophic factors
Epidermal growth factors
Fibroblast growth factors
Hepatocyte growth factor
Heregulin
Glial growth factors
Insulin-like growth factors I, II
Nerve growth factors
Platelet-derived growth factor
Transforming growth factors <ga>, <gb>1---5
Vascular endothelial cell growth factors
Growth inhibitors
Leukemia inhibiting factors
Oncostatin M
Mullerian inhibiting substance
Transforming growth factor <ga>, <gb>1---5
Tumor necrosis factor
Chemotactic factors
Complement (C3)
Monocyte chemotactic protein-1
Macrophage inflammatory protein 1 <ga>, <gb>
MIF
RANTES
CSF, colony-stimulating factor; GM-CSF, granulocytemacrophage colony-stimulating factor; IL-3, interleukin-3, MIF = macrophage migration inhibitory factor; RANTES = regulated on activation, normal T cell expressed and secreted.
TH1 lymphocytes express inflammatory cytokines that are involved in the effector functions of cell-mediated immunity. The primary cytokines of TH1 lymphocytes are interferon-γ (IFN-γ) and interleukin (IL)-2. TH2 lymphocytes produce cytokines such as IL-4 and IL-10, which often antagonize the inflammatory effects of IFN-γ and stimulate B-cell differentiation. IL-4 is required for the production of antibodies, IgA, and IgE, primarily associated with mucosal surfaces as this cytokine stimulates isotype switching to the α and ε heavy chains. TH1 lymphocytes appear to be more important in the defense against intracellular pathogens, whereas TH2 lymphocytes have an important role in the protection against parasitic disease. These two cell populations also are responsible for different pathologic states with TH1 responses involved in organspecific autoimmune disorders, including experimental autoimmune uveoretinitis, and TH2 cells involved in atopic disease.7
CD8+ lymphocytes are primarily responsible for cytolysis of virus-infected cells, malignant cells, and tissue allografts. This type of cell killing is highly specific as antigenic peptides are presented to the CD8+ cell in the context of class I MHC molecules and require direct contact of the CTL with the target cell. CD8+ cells also participate in cell-mediated immunity by activating macrophages. The role of CD8+ cells as “suppressor” cells is less clear, although a distinct subpopulation of non-CTL CD8+ cells may be distinguished.
Mononuclear Phagocytes
Cells of the mononuclear phagocyte system are derived from a common myeloid progenitor cell in the bone marrow. Their primary functions are phagocytosis and antigen presentation. In the peripheral blood, they are found as incompletely differentiated cells known as monocytes. Once in tissue, they mature and are known as macrophages.
Macrophages function as important effector cells of innate immunity. They are capable of phagocytizing foreign particles and microorganisms and killing them with lysosomal enzymes. They also produce and secrete cytokines that recruit other inflammatory cells and promote the inflammatory response. Among the cytokines typically expressed by macrophages are tumor necrosis factor (TNF)-α, IFN-γ, IL-1, and IL-6.
Macrophages also act as efficient antigen-presenting and costimulatory cells for T cells. T cells, in turn, may secrete cytokines, especially IFN-γ, which activate macrophages, upregulating their killing ability and antigen-presenting capacity.
Dendritic Cells
Dendritic cells are extremely efficient antigen-presenting cells with distinctive spinelike projections. Interdigitating dendritic cells are bone marrow derived and are found in the interstitium of most organs. In the skin and conjunctiva, they are known as Langerhans cells and bear the CD1 cell surface marker. Langerhans cells are extremely mobile, migrating from the skin to the peripheral lymph nodes, and are particularly effective at presenting antigen to CD4+ cells. Follicular dendritic cells are found in the germinal centers of lymph nodes, the spleen, and the mucosal lymphoid tissue. They are unrelated to interdigitating dendritic cells but also are extremely important antigen-presenting cells within lymphoid tissue.1
Neutrophils
Leukocytes containing abundant cytoplasmic granules play vital roles in the elimination of microorganisms and in the acute inflammatory response. Neutrophils comprise more than 90% of granulocytes and between 60% and 70% of the circulating leukocyte pool. They are extremely mobile, responding rapidly to chemotactic stimuli, adhering to specific ligands on the endothelial lining of blood vessels, and, via diapedesis, extravasating into tissue sites of injury. Chemoattractants for neutrophils include the byproducts of bacterial metabolism; C5a, which is a byproduct protein component of the complement cascade system; as well as leukotriene B4 and platelet- activating factor (PAF), both derived from cell membrane lipids.8
Various cellular processes including adhesion, migration, proliferation, differentiation, and activation are modulated by cell adhesion molecules (CAMs). Adhesion molecules are expressed on leukocytes, vascular endothelium, and epithelial cells and have been identified within various structures of the eye including the cornea, conjunctiva, choroid, uvea, and optic nerve.9 They are grouped in four major families: the integrins, the selectins, the cadherins, and the immunoglobulin supergene family.10 Endothelial CAMs important in regulating the infiltration of neutrophils into sites of inflammation include E-selectin and intercellular adhesion molecule-1 (ICAM-1). ICAM-1 binds to a complement receptor known as membrane attack complex type 1 (MAC-1) (i.e., complement receptor 3 or CR3) on the neutrophil, establishing firm adhesion and leading to extravasation.11 ICAM-1 also plays a role in ocular hypersensitivity reactions as reflected by its increased expression in atopic patients during allergen-specific conjunctival challenge.11,12
During phagocytosis, neutrophils engulf invading microorganisms, isolating them in phagosomes. Cytoplasmic granules containing a variety of cytotoxic enzymes fuse with the phagosome and destroy the pathogen. Most invading microorganisms are coated, or opsonized, with IgG or another complement protein byproduct known as iC3b. These can then bind to specific receptors on neutrophils (FcRII and MAC-1), facilitating phagocytosis.
The neutrophil has both oxygen-dependent and oxygen-independent antimicrobial systems. The oxygen-dependent system produces reactive oxygen metabolites (“respiratory burst”) that are toxic to microorganisms. The oxygen-independent system relies on the cationic peptides and enzymes released from cytoplasmic granules for killing.
Eosinophils
Eosinophils are important effector cells in atopic disease and parasitic infection. They contain a bilobed nucleus and distinctive acidophilic granules and are far more prevalent in tissue than in peripheral blood. Their cytoplasmic granules contain a number of basic proteins, including major basic protein (MBP), eosinophil cationic protein (ECP), eosinophil-derived neurotoxin (EDN), and eosinophil peroxidase (EPO), that are responsible for the eosinophils uptake of acid stains. MBP makes up more than 50% of total granule protein, is known to be extremely toxic to parasites, and is a vital component of eosinophil ADCC. MBP also is responsible for damage to epithelial cells, as seen in the respiratory epithelium in patients with asthma, and which is similar to the damage in the corneal epithelium in patients with vernal and atopic keratoconjunctivitis.
IL-5, produced by the TH2 subset of CD4+ cells and activated mast cells, is a potent stimulant for eosinophil production and activation. It also prolongs eosinophil survival and causes secretion of granule proteins. IL-4, another TH2 cytokine important in immediate hypersensitivity disease, causes endothelial cells to upregulate the expression of the adhesion molecule vascular cellular adhesion molecule (VCAM-1), which binds to its ligand VLA-4 on eosinophils. This results in eosinophil recruitment to sites of immediate and chronic immunologic hypersensitivity inflammatory reactions.
Mast Cells and Basophils
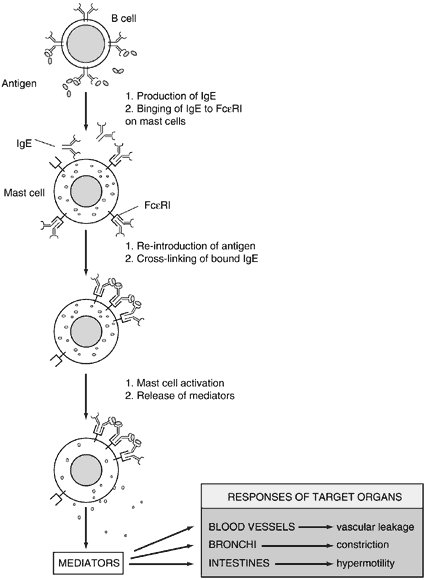
Mast cells and basophils play important roles in mediating immediate hypersensitivity. They each express large numbers of high-affinity IgE receptors (Fcε RI) on their cell surfaces. Mast cells are localized to mucosa, epithelial surfaces, and connective tissue. Basophils may be considered the peripheral blood counterpart to the mast cell.
Mast cells can be divided into two types based on their expression of secretory granules: tryptase mast cells (MCT) and tryptase-chymase mast cells (MCTC). MCT are found primarily in the lung and intestinal mucosa, whereas MCTC are localized to the skin and intestinal submucosa. The predominant form of mast cell found in the normal conjunctiva are MCTC; however, there is a noticeable increase in the MCT type in chronic conjunctival inflammatory conditions. It is estimated that more than 50 million mast cells are present in the conjunctiva.
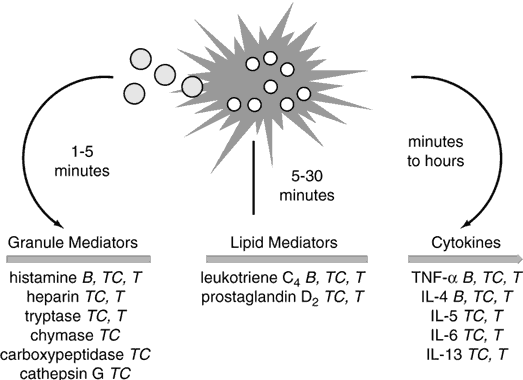
Mast cells and basophils are activated by the cross-linking of FcεRI molecules on their surface after the binding of multivalent antigen to sufficient IgE. Activated mast cells release their preformed mediators in a regulated fashion and then synthesize lipid-derived mediators of inflammation.
The prototype preformed vasoactive mediator released by mast cells is histamine. Histamine binding to its cell receptors results in vasodilation, endothelial cell contraction, and associated plasma leakage, as well as bronchial and intestinal smooth muscle constriction. Other preformed mediators include tryptase, chymase, carboxypeptidase-A, proteoglycans, eosinophil chemotactic factor, and neutrophil chemotactic factor. Lipid-derived mediators synthesized after activation include leukotrienes B4 (one of the most potent neutrophil chemoattractants known to the human body), C4, D4, and E4 (also known as SRS-A, a slow-releasing substance of anaphylaxis), prostaglandin D2, and PAF, which all have vasodilatory and smooth muscle constrictive effects (Fig. 3).4
|
Lymphoid Tissues
Organized lymphoid tissue allows for the concentration of lymphocytes and antigen-presenting cells at anatomically defined sites. The eye is immunologically unique in that it has no formed lymph nodes in the orbit or associated with the lacrimal gland, eyelids, or conjunctiva. Lymphocytes normally reside in the substantia propria of the acini of the lacrimal gland and the conjunctiva.
Lymph nodes situated along lymphatic pathways throughout the body provide a mechanism for the host to survey for foreign molecules. Each lymph node is divided into an outer cortex, which contains lymphoid follicles and is densely packed with cells, and the more sparsely populated medulla, which contains the vascular and lymphatic sinusoids. Follicles are B-cell zones and may develop germinal centers, where activated B cells proliferate and interact with follicular dendritic cells.
T cells are localized between the follicles and the deep cortex in the parafollicular areas. These cells are primarily CD4+ and interact with the interdigitating dendritic cells. The anatomic structure of the lymph node provides areas of interaction for B cells, T cells, and antigen-presenting cells. This structure expands or regresses in response to variable antigen exposure.
The spleen has an anatomic structure similar to that of the lymph node, is the primary site of immune response to blood-borne antigen, and appears to be the primary lymphoid site for intraocular antigenic stimulation. The white pulp of the spleen contains densely packed lymphoid tissue. T cells are localized to the periarteriolar lymphatic sheaths and B cells to the follicles and germinal centers.1
Other lymphoid tissue is found as aggregates of lymphocytes and accessory cells in the submucosa of the gastrointestinal and respiratory tracts. The skin contains lymphocytes and Langerhans cells in the dermis and epidermis.
Immunoglobulins
Immunoglobulins, or antibodies, are the glycoprotein products of antigen-stimulated B cells. The only function of specialized, terminally differentiated B cells, called plasma cells, is the production and secretion of large amounts of immunoglobulin, which is found in both membrane-bound (serving as B-cell surface receptors) and soluble forms. They are widely distributed in plasma and secretory fluids such as tears, milk, and mucous.
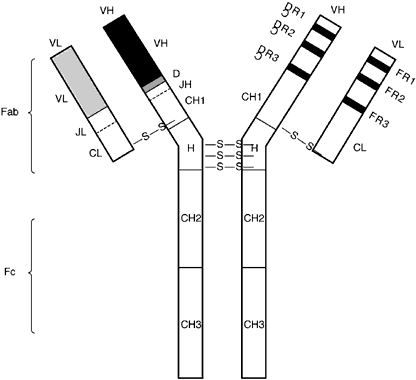
All immunoglobulins have the same basic structure with two identical heavy chains combining with two identical light chains. The two heavy chains are bound together by disulfide bonds, and each light chain is similarly attached to each heavy chain. Despite this similarity in basic structure, immunoglobulins may be divided into different classes based on certain physiochemical characteristics. The common physiochemical and antigenic properties of each class are based on shared regions of heavy-chain amino acid sequences. There are five basic classes, or isotypes: IgM, IgD, IgG, IgA, and IgE. There also are further subclasses: IgG1, IgG2, IgG3, IgG4, and IgA1 and IgA2. Different isotypes and subtypes mediate distinct effector functions. The two light-chain types, δ and μ, do not confer any effector function.
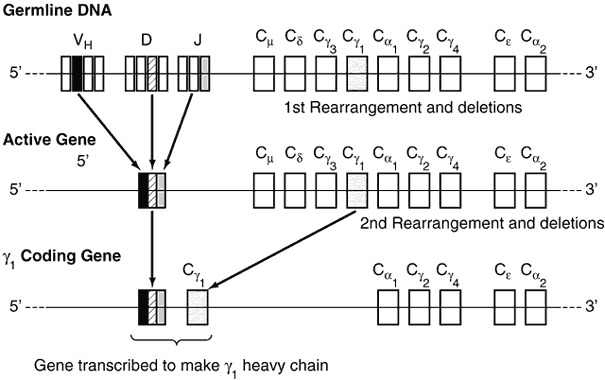
The diversity of the immunoglobulin repertoire results from the remarkable variability of the antigen-binding sites. These binding sites are composed of three hypervariable regions of both the light and heavy chains (Fig. 4). The variability in these regions is a result of somatic genomic recombination, multiple germline genes, junctional diversity, and somatic mutations (Fig. 5). The first immunoglobulin molecules expressed on mature B cells are IgM and IgD. IgD is almost exclusively membrane bound, contributing to less than 1% of total plasma immunoglobulin. Its function remains unclear.
|
|
IgM plays a significant role in the primary immune response. Antigen contact with membrane-bound IgM initiates cell division, the production of secreted immunoglobulin, and the formation of memory B cells. In its secreted form, IgM usually is found as a pentamer, allowing for multiple contacts with antigen. It is predominantly confined to the intravascular pool and accounts for approximately 10% of immunoglobulins.
IgG is the most abundant immunoglobulin in normal human serum, accounting for approximately 75% of the total immunoglobulin pool. It is the primary mediator of immunoglobulin effector functions. IgG subclasses (except for IgG4) activate the classical pathway of the complement system. IgG helps mediate phagocytosis, ADCC, and cytokine secretion through different Fc receptors on a variety of cell types. IgG also is the only immunoglobulin to cross the placenta, thus conferring passive maternal immunity to the neonate. Thus, IgG ocular disorders are the only ones that the mother can transplacentally “passively” transfer to the infant at birth, such as myasthenia gravis.
Although thought to be a relatively unimportant component of systemic humoral immunity, IgA plays a major role in mucosal immunity. Dimeric IgA binds to specific Fc α receptors, “secretory components,” on epithelial cells of organs such as the intestine, as well as on the conjunctival surface. The secretory component shuttles the dimeric IgA through the cell until it is cleaved at the luminal side. Hence, dimeric IgA enters the mucosal lumen where it can neutralize pathogens. IgA is the predominant immunoglobulin in tear fluid, milk, saliva, and tracheobronchial secretions.1
IgE usually is found in small amounts in the serum of normal individuals but may be increased greatly in patients with atopic disease. It is responsible for immediate hypersensitivity reactions and for immunity to parasites. CD4+ TH2 cells produce IL-4, which promotes the production of IgE. IgE with specific epitopes to allergens has been isolated in the tear fluid of patients with atopic disorders (e.g., ragweed-specific IgE) in ragweed-sensitive patients.4
T-Cell Receptor Complex
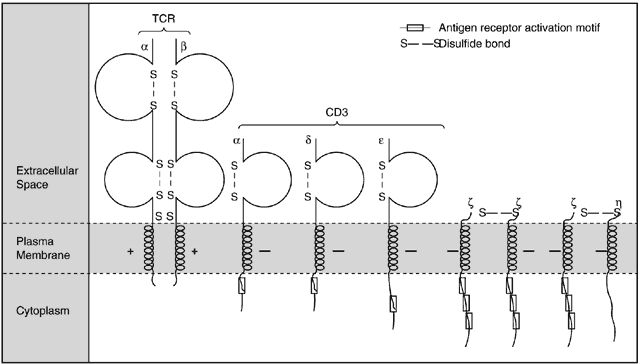
Antigen recognition by T cells is accomplished by specific TCR binding of antigenic peptide presented in the context of self-MHC molecules on antigen-presenting cells. TCR molecules do not recognize soluble antigens, native protein, or nonproteins as antigens. The TCR is a heterodimer of two polypeptide chains, α and β or γ and δ, linked by disulfide bonds. More than 90% of T cells in human peripheral blood, lymphoid tissue, and normal conjunctiva are TCR-α/β-positive.13 Both chains have variable and constant regions. The variable region shares many amino acid residues and a common tertiary structure with immunoglobulins. TCR molecules share enough sequence homology with immunoglobulin molecules to be considered members of the immunoglobulin supergene family.
As with immunoglobulins, the diversity of the TCR repertoire results from variability at the antigen-binding sites. This variability is a result of mechanisms that fundamentally resemble those for the immunoglobulin repertoire: multiple germline genes, somatic genomic recombinations, and junctional diversity. However, there is no evidence that somatic mutations result in a change of function or affinity of the TCR.
The TCR is noncovalently associated on the T-cell surface with CD3 and other proteins to form a functional TCR complex. The CD3 and these other proteins act as substrates for tyrosine kinases and actually function as the signal-transducing component for the TCR complex (Fig. 6).1
|
The Major Histocompatibility Complex
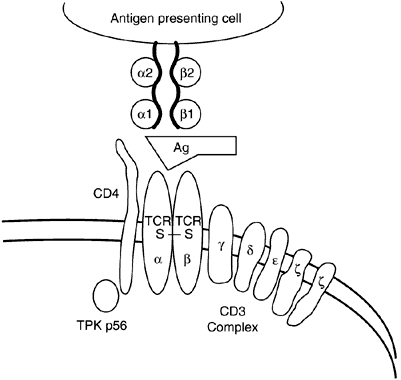
The MHC is a large, polymorphic genetic region located in humans on chromosome 6. It codes for a number of cell surface proteins, designated class I and II molecules, critical for distinguishing self from nonself. The MHC was discovered as the major antigenic system responsible for the rejection of tissue grafts from one individual to another (allografts). In humans, the MHC is known as the human leukocyte antigen (HLA) region based on the original demonstration of these proteins in humans on leukocytes. Class I MHC genes encode HLA-A, -B and -C surface antigens and are expressed on all nucleated cells. Class II genes encode HLA-D-related proteins (i.e., DR, DP, DQ) found on macrophages, other antigen-presenting cells, and B cells.
Class I and II molecules are responsible for mediating the interactions of T cells and antigen-presenting cells. CD4+ cells only recognize antigenic peptides in the setting of self MHC class II molecules (Fig. 7), whereas CD8+ cells only recognize peptides in the setting of self MHC class I molecules (Fig. 8). The peptide fragments that bind class II molecules generally are of extracellular origin, whereas endogenously derived peptides (viral particles) are bound by class I molecules.
|
|
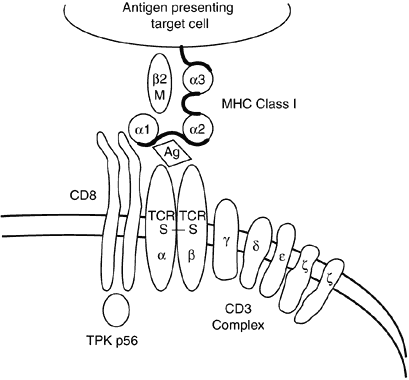
Class I and II MHC products bind short stretches of peptides (class I, 9 to 11 amino acids in length; class II, 10 to 30 amino acids in length). The polymorphic amino acid residues in the peptide-binding region of the MHC molecules are responsible for both peptide binding and binding to the complementary ligand on the T cell.1,8
The Complement System
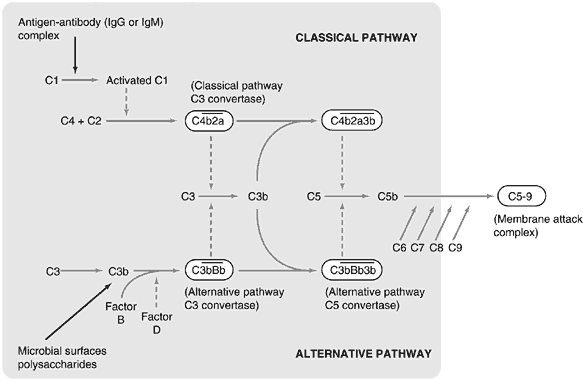
The complement system is a collection of functionally related proteins that interact with each other in a tightly regulated fashion to play a major role in host defense. A characteristic of the system is the sequential activation of proteolytic enzymes that allows for dramatic amplification of the activated molecules. Its principle functions are lysis of bacterial cell walls and enveloped viruses, opsonization, activation of the inflammatory response, and the solubilization and clearance of immune complexes.
Direct lysis of invading cells is mediated by the formation of the membrane attack complex (MAC) by terminal components of complement. The MAC inserts into the lipid bilayer of cell or viral membranes to form pores that allow for the rapid influx of water into the cell and consequent osmotic lysis.
Opsonization of microorganisms results from the binding of complement proteins C3 and iC3b to their surfaces. Neutrophils and macrophages express receptors for these proteins (CR1 to bind C3, MAC-1 and CR4 to bind C3bi), and thus phagocytosis of the opsonized pathogen is enhanced.
Activation of inflammation by complement is mediated by proteolytic fragments of early complement proteins (C3a, C4a, and C5a) termed the anaphylatoxins (Fig. 9). Anaphylatoxin binding to mast cell and basophils results in degranulation and the release of histamine and other vasoactive mediators as noted above. C5a also can act directly on vascular endothelium to cause contraction, vascular leak, and exocytosis.
|
Large numbers of immune complexes may be formed in the setting of a vigorous immune response to high levels of circulating antigens. Their deposition in blood vessel walls can activate complement, resulting in inflammatory reactions that may damage local tissue. Complement binding to the Fc region of immunoglobulin molecules sterically inhibits the formation of immune complexes. The complement system also promotes the clearance of immune complexes from the circulation by the mononuclear phagocyte system.
The complement system contains two pathways, the classical and the alternative, that converge on the central complement protein C3. Complement activation along the classical pathway is initiated by antigen-antibody complexes, whereas activation along the alternative pathway occurs on microbial surfaces in the absence of antibody. Both pathways use C3 to produce enzymes that result in the formation of the MAC. Downregulation of the system is mediated by various soluble and membrane-bound proteins at steps throughout the cascade.1,4,14
Tolerance
Successful immune regulation requires mechanisms to generate specific immunologic self unresponsiveness, or tolerance. Tolerance to self is a learned process during which self-reactive lymphocyte clones are either prevented from developing or are inactivated after recognizing self-antigen. Loss of tolerance results in autoreactivity and, potentially, autoimmune disease.
The induction of self-tolerance in immature lymphocytes occurs in the generative lymphoid organs (i.e., the thymus for T cells and the bone marrow for B cells) and is termed central tolerance. Immature lymphocytes are more susceptible to the induction of tolerance than mature lymphocytes. If these cells encounter and recognize antigens before developing functional competence, they are deleted (clonal deletion) or rendered incapable of immunologic response (clonal anergy). The antigens encountered in the thymus and bone marrow are predominantly self-antigens, and thus the mature lymphocytes that emerge from these organs are depleted of self-reactive clones. Central tolerance in T cells results from clonal recognition of self-peptide-MHC complexes leading to either deletion or anergy in a process termed negative selection.1
Any self-reactive lymphocyte clones that escape this process and move into peripheral tissue subsequently may be inactivated as they encounter self-antigen under conditions that favor tolerance rather than immune activation. Conditions that influence this process, termed peripheral tolerance, include the type, amount, and portal of entry of antigen, the presence of competent antigen-presenting cells, and the type of responding lymphocyte.
The activation status of tissue antigen-presenting cells is a crucial determinant of peripheral tolerance. CD4+ cells that recognize antigen presented by antigen-presenting cells that cannot deliver a costimulatory signal (e.g., the absence of HLA B7 on a macrophage) become anergic. This period of anergy lasts for weeks, during which these cells will be unable to secrete IL-2 to signal autoproliferation. In addition, some antigen-presenting cells provide a direct inhibitory signal. CD4+ cells become unresponsive if antigen is presented to them by other class II MHC-bearing cells. The local cytokine milieu also influences peripheral tolerance. For example, T cells exposed to high concentrations of IL-2 at the time of antigen exposure die.
Peripheral B-cell tolerance occurs through similar mechanisms. B cells binding to antigen in the absence of T-cell help do not result in the production of antibody. In general, compared with the induction of anergy in T cells, B-cell anergy is shorter lived and requires higher concentrations of antigen to elicit.1
Transplantation immunologists have long recognized that certain sites, namely the anterior chamber of the eye, the brain, and the testes, are privileged in their ability to accept allografts. Immune privilege in the eye seems related to three factors: (1) the nature and function of ocular antigenpresenting cells; (2) the integrity of the blood-ocular (brain) barrier; and (3) the presence of an immunomodulatory intraocular environment. A unique form of systemic immune deviation exists after the inoculation of antigen into the anterior chamber, termed anterior chamber associated immune deviation (ACAID). In a simple experiment, the iris, ciliary body, and cornea were identified as secreting factors associated with the development of ACAID. The dendritic cell appears to be the messenger cell that is able to migrate via the aqueous humor through the trabecular meshwork into the blood and travel preferentially to the spleen. It appears that antigen is then presented in the context of class I MHC rather than the more usual class II MHC molecules. This induces a systemic suppressive response as reflected by depressed antigen-specific delayed-type hypersensitivity and an increase in the generation of cytotoxic T cells. It is believed that ACAID represents a physiologic adaptation to limit nonspecific inflammation and “bystander” cell injury related to delayed-type hypersensitivity, thus preserving the anatomically sensitive visual axis.15–17
Autoimmune disease results from the failure to maintain self-tolerance. This failure may result from the abnormal selection of self-reactive clones, the stimulation of lymphocytes that typically are anergic to self-antigen, or from the release of self-antigen from sites previously inaccessible to immune surveillance. Autoimmune disease also may develop when lymphocytes responding to foreign antigen recognize and cross-react to a self-protein in a process termed molecular mimicry.1