DEVELOPMENTAL BIOLOGY91
Eye development becomes evident at approximately 22 days gestation, when the optic sulci appear as bilateral grooves in the neural ectoderm of the embryo. At approximately 25 days gestation, as the rostral neural tube fuses, the neural ectoderm evaginates further to form the optic vesicles. The optic vesicles are attached to the developing brain via the optic stalks. As the optic vesicles grow laterally and approach the surface ectoderm, they invaginate to form a bilayered optic cup. The inner and outer layers of the optic cup proceed to form the neural retina and the retinal pigmented epithelium, respectively. The invaginated optic cup forms a somewhat spherical structure that is open anteriorly and inferiorly. This inferior “seam” is alternatively called the optic fissure, the choroidal fissure, or the embryonic fissure, and it transmits the hyaloid vascular system beginning at approximately 5 weeks gestation. At 6 weeks gestation, the optic fissure “zippers” shut, beginning equatorially and proceeding anteriorly and posteriorly.
ANOPHTHALMIA
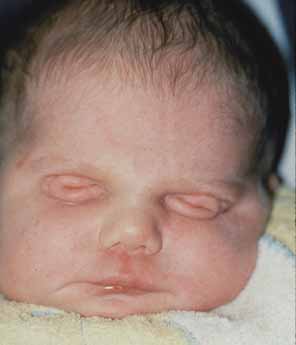
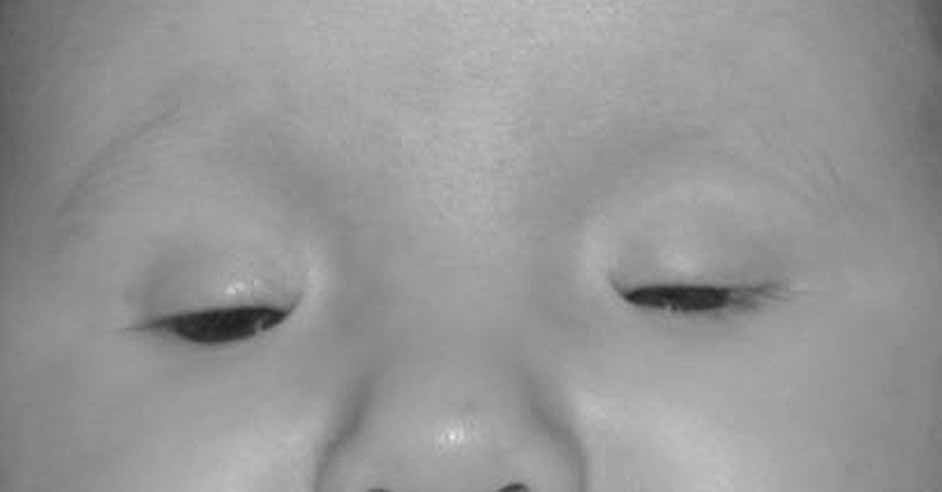
Anophthalmia refers to the complete absence of ocular tissue, presumably caused by abnormalities of optic vesicle formation or maturation. Patients may have unilateral or bilateral anophthalmia, and generally have short palpebral fissures and small orbits. In many cases when there is no clinically apparent ophthalmic tissue, remnants of lens epithelium, neuroretinal, and fibrovascular, choroid-like tissue can be detected on histologic sectioning. Terms such as “true anophthalmia,” “extreme microphthalmia,”10 and “clinical anophthalmia”11 have been used in the literature to describe what may be, in truth, a phenotypic continuum between anophthalmia and microphthalmia (see later).
Anophthalmia may be isolated or associated with a broader syndrome (Table 2).12–19 Isolated anophthalmia is generally an autosomal recessive condition.20 Driggers et al. reported a child with isolated bilateral anophthalmia and an apparent balanced chromosomal translocation {46,XX,t(3;11)(q27;p11.2)}.21 Fantes et al. found that this child had, in fact, a submicroscopic deletion that eliminated the SOX2 gene on chromosome 3, and went on to show nonsense mutations in SOX2 in four of 11 subjects with bilateral anophthalmia.22 SOX2 lies within an intron of a nonexpressed gene, SOX2OT, and is expressed in neuroectoderm early in development.23 SOX2 protein interacts cooperatively with PAX6 in the induction of lens development and delta-crystallin expression.24 Hagstrom, Traboulsi et al. found a null mutation in SOX2 in a girl with bilateral clinical anophthalmia and absence of the optic nerves, chiasm, and optic tracts (unpublished data)
TABLE 2. Clinical Syndromes Associated with Anophthalmia
| Syndrome | OMIM # | Inheritance | Characteristics in Addition to Anophthalmia |
| Fryns | 60076 | AR | Orofacial clefting, uterine abnormalities, ear abnormalities, neural tube defects |
| Oculocerbral-cutaneous | 164180 | AD | Orbital cysts, focal dermal hypoplasia, cerebral malformations, cleft lip/palate |
| Lenz microphthalmia | 309800 | X-linked | Microphthalmos, mental retardation, distal limb abnormalities, microcephaly, orofacial clefting, tooth & skeletal anomalies, hearing loss, GU malformations, imperforate anus |
| Matthew-Wood | 601186 | Sporadic | Pulmonary hypoplasia |
| Waardenburg | 206920 | AR | Syndactyly/other distal limb abnormalities, mental retardation, skeletal anomalies |
| 14q22–23 del | 607932 | Sporadic | Polydactyly, pituitary hypoplasia, ?SIX6 hemizygosity |
| 605856 | Growth & mental retardation, callosal agenesis, heminasal hypoplasia, atypical clefting, external ear abnormalities, | ||
| 60092 | Sporadic | Esophageal atresia |
Syndromes associated with anophthalmia. Clinically, phenotypic variability exists. Abbreviations: OMIM, online Mendelian inheritance in man; AR, autosomal recessive. References and specific cases may be found in OMIM.
Reconstructive surgery can be helpful in patients with congenital anophthalmia.25 Orbital expansion can be achieved with spherical implants, orbital osteotomies, bone grafts, and/or orbital expanders. Conjunctival sac reconstruction is achieved using serial expanders and buccal mucous membrane grafts. Lid reconstruction sometimes requires tissue flaps and skin grafts.
MICROPHTHALMIA
A microphthalmic eye is one with axial length less than two standard deviations below the age-adjusted mean (ie, <19 mm in a 1-year-old child and <21 mm in an adult.) Microphthalmia encompasses a range of phenotypes. Specific terms, outlined later, are applied to subgroups of microphthalmic eyes with certain characteristics. Authors of scientific communications may not use the same classification scheme or may use the more general term “microphthalmia” when a more specific term could apply. Although we will present these terms as distinct entities, remember that they likely represent points along a single, phenotypic continuum.
Simple Microphthalmia/Nanophthalmos
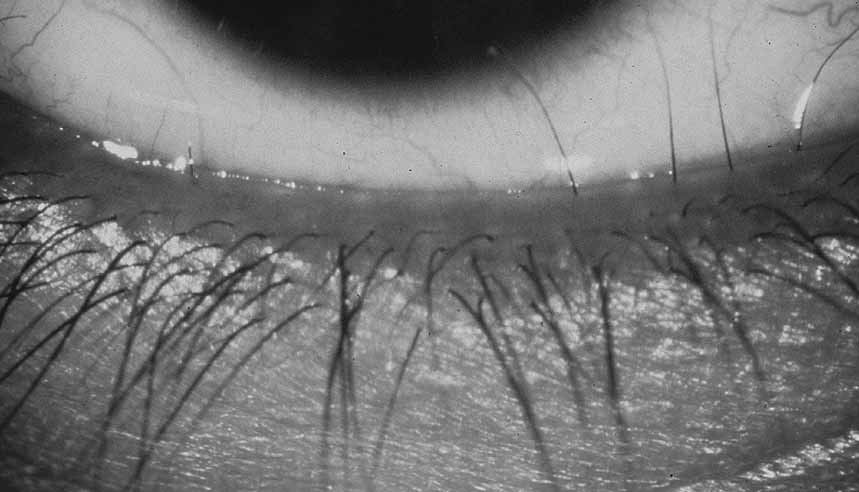
Simple microphthalmia refers to a small but structurally normal eye.26 The term “nanophthalmos” is used synonymously, but especially when such eyes develop angle-closure glaucoma caused by crowding from the normal-size (ergo, disproportionately large) lens or uveal effusions. Weiss et al. noted that, in general, the posterior segment is proportionately shorter than the anterior segment in such patients.26 Simple microphthalmia/nanophthalmos can be inherited as part of a broader syndrome (Table 3)27–29 or as an individual trait. The sclera of nanophthalmic eyes consists of irregularly arranged collagen lamellae, absence of normal elastic fibers, and abnormal glycogen-like deposits.30 These changes likely result in the increased thickness and decreased elasticity of the nanophthalmic sclera—changes that result in decreased blood flow through the vortex veins and/or decreased transscleral flow of protein. This alteration in ocular fluid dynamics increases the risk of uveal effusion and choroidal detachment, especially after surgery.31–33
TABLE 3. Syndromes With Simple Microphthalmos/Nanophthalmos as a Phenotype
| Syndrome | OMIM# | Inheritance | Locus (Gene) | Characteristics in Addition to Simple Microphthalmia |
| Kenney-Caffey | 127000 | AD vs X-linked | Unknown | Short stature, dense tubular bones, narrow marrow cavities, hypocalcemia/phosphatemia |
| Mucolipidosis IIIa | 252600 | AR | 4q21-q23 (N-acetyl-glucosamine–1-phosphotransferase) | Short stature, cloudy corneas, retinopathy, bone abnormalities, aortic insufficiency, mental retardation, skin thickening |
| 252605 | ||||
| Oculodentodigital Syndrome | 164200 | AD | 6q21–23.2 (Connexin 43) | Characteristic facies, microcephaly, syndactyly, hypo/aplasia middle phalanges, dental abnormalities, glaucoma, cataract, optic atrophy |
AD, autosomal dominant; AR, autosomal recessive. Clinically, phenotypic variability exists.
aMucolipidosis type III includes three biochemical complementation groups. The ophthalmic literature does not specifically differentiate these, but the “pseudo-Hurler” phenotype is consistent.
The precise embryologic mechanism for simple microphthalmia/nanophthalmos is unclear. Based on finding this phenotype in patients with fetal alcohol syndrome, achondroplasia, and myotonic dystrophy, Weiss et al. postulated that decreased size of the optic cup, altered vitreous proteoglycans, low intraocular pressure, and abnormal release of growth factors may be important in the pathogenesis of simple microphthalmos.26 These authors point out that given many of these patients have normal corneal diameters and that the area primarily affected is the posterior segment, the pathogenesis in many of these conditions may be related to postnatal, rather than prenatal, changes in ocular development and growth.
In 1998, Othman et al. described a pedigree with autosomal dominant nanophthalmos that localizes to a 14.7cM region on chromosome 11 (NNO1).34 Affected family members had high hyperopia (range of +7.25 to +13 D) and short axial lengths (range of 17.55–19.28 mm). Twelve of 22 affected family members had a history of angle-closure glaucoma or had occludable angles. Although OMIM lists a second nanophthalmos locus, NNO2, on 15q12-q15, the phenotype described in this pedigree is more consistent with a colobomatous, complex microphthalmia (see later).35
Complex Microphthalmia
In cases in which the eye is not only small but also abnormal in other respects, the term “complex microphthalmia” is applied. Examples of associated findings are microcornea, sclerocornea, corneal opacities, aniridia, Peters anomaly and cataract. Complex microphthalmia can be associated with a syndrome (Table 4)19,35–61 or can occur as an isolated trait. Complex microphthalmia encompasses a diversity of phenotypes. Patients in the same family with the same underlying disorder may have phenotypes of varying severity. For example, patients with the Lenz microphthalmia syndrome (OMIM 309800) may have anophthalmia, colobomatous microphthalmia, or noncolobomatous microphthalmia.62
TABLE 4. Syndromes with Complex Microphthalmia as a Phenotype
| Syndrome | OMIM # | Inherit | Locus (Gene) | Features in Addition to Complex Microphthalmia |
| Aicardi syndrome | 304050 | XLD | Xp22 | Agenesis of corpus collasum; chorioretinal abnormality; infantile spasms; microcephaly, CL/CP; rib/vertebrae abnormalities; brain abnormalities; neoplasia; precocious puberty |
| Arhinia, choanal atresia, & microphthalmia | 603457 | AD? | Unknown | Complete absence of nose, choanal atresia, cleft palate |
| BOFS (Branchio-oculo-facial syndrome) | 113620 | AD | Unknown | Growth def. (pre/postnatal); microcephaly; micrognathia; coloboma; ear abnormalities; cataract; CL/CP; dental anomalies; branchial anomalies |
| CATM | 156850 | AD | 16p13.3 | Congenital cataract |
| CHARGE | 214700 | AD, sporad | Unknown | Coloboma, hear anomaly, choanal atresia, retardation of growth & development, genital & ear abnormalities |
| COFS (Cerebro-oculo-facio-skeletal syndrome) | 214150 | AR | 19q13.2-q13.3 (XPD); 13q33; 10q11 (ERCC6) | Neurogenic arthrogryposis; cataract; microcephaly; brain malformation; blepharophimosis; hirsutism; kyphoscoliosis; osteoporosis; renal defects; large pinna; wide-spaced nipples; verticle talus; flexion contractures |
| Coloboma-obesity-hypogenitalism-mental retardation syndrome | 601794 | AD | Unknown | Cataract, obesity, hypogonadism, MR |
| Dextrocardia with unusual facies | 221950 | AR | Unknown | Dextrocardia, dysmorphic facies, folding on foot, normal growth |
| Facio-oculo-auriculo-vertebral dysplasia (Goldenhar) | 164210 | Sporad AD | 14q32 | Unilateral deformity of external ear w/ small ipsilateral half of face; epibulbar dermoid; vertebral anomalies; CL/CP; deafness; CN VII palsy; epibulbar dermoids; lid colobomas; Duane's syndrome |
| Facio-thoraco-genital syndrome | 227320 | AR | Unknown | Anteverted nares; long philtrum; thin upper lip; pectus excavatum; micrognathia; genital abnl.; wide great toes; thumbs; hypoplastic nails |
| Focal Dermal Hypoplasia (Goltz-Gorlin syndrome) | 305600 | XLD (lethal in males) | Xp22 | Atrophy & linear pigmentation of skin; herniation of fat through dermal defects; multiple papillomas of mucous membranes; colobomas; digital anomalies; hypoplastic teeth; MR; striated bones |
| Frontonasal dysplasia sequence | 136760 | Sporad | Unknown | Hypertelorism; lateral displacement of inner canthi; widow's peak; deficit in midline frontal bone; nasal tip abnormalities; absence of corpus callosum |
| GOMBO syndrome | 233270 | AR | 3p or 22q ? | Microcephaly; brachydactyly w/ clinodactyly 5, delayed growth at puberty; MR; oligophrenia (affected w/ 46, X,Y, ish der(3),t(3,22)(p25;q13)) |
| Hallermann-Streiff syndrome (Francois dyscephalic syndrome) | 234100 | Sp. | Cataracts; coloboma; small, pinched nose; hypotrichosis; postnatal growth def.; brachycephaly; malar hypoplasia; dental abnl.; atrophy of skin; wormian bones; thin, gracile long bones | |
| Kapur-Toriello syndrome | 244300 | AR | Unknown | Coloboma, MR, congenital heart defect, CL/CP, malrotation of intestine; displaced kidneys |
| Lenz microphthalmia syndrome | 309800 | X-linked | Xq27-q28 Xp11.4-p21.2 | MR, distal limb abnormalities, microcephaly, oro-facial clefting, tooth & skeletal anomalies, hearing loss, GU malformations, imperforate anus |
| Macrosomia w/ microphthalmia, lethal | 248110 | AR | Unknown | Macrosomia, median clefting, early infant death from overwhelming infection |
| Meckel syndrome, type 1–3 | 24900 | AR | 17q22-q23, 8q24, 11q13 | Renal cysts; developmental anomalies of the CNS (esp. encephalocele); hepatic duct dyplasia & cysts; polydactyly |
| Microphthalmia w/ cyst, limb anomalies, bilateral facial clefts | 607597 | ? | Unknown | Features similar to Waardenburg ophthalmo-acromelic syndrome (206920), cerebro-oculonasal syndrome (605627), and craniotelencephalic dysplasia (218670), but not complete for any. |
| MIDAS syndrome | 309801 | X-linked | Xp22.31 | Dermal atrophy, linear skin defects; sclerocornea, orbital cysts, presumed lethal in hemizygous males |
| MMEP | 601349 | AD (?) | 6q21 (SNX3 {sorting nexin 3}, 1 case) | Microcephaly, ectrodactyly of the lower limbs, prognathism |
| Nanophthalmos 2 (NNO2) | 605738 | AD | 15q12-q15 | Coloboma, microcornea. Variable expressivity w/ some showing only Peters anomaly, optic nerve agenesis |
| Oculodentodigital Syndrome | 164200 | AD | 6q21–23.2 (Connexin 43) | Characteristic facies, microcephaly, syndactyly, hypo/aplasia middle phalanges, dental abnormalities, glaucoma, cataract, optic atrophy |
| Oculo-dento-osseous dysplasia | 257850 | AR | Unknown | Microcornea; dysplastic iris; PHPV; long, narrow nose w/ hypoplastic nasal alae; malformed teeth w/ abnormal enamel; 4–5 syndactyly (fingers); widened long bones |
| Oculo-facio-cardio-dental syndrome | 300166 | XLD ? male lethal | Unknown | Congenital cataract; long, narrow face; CL/CP; ASD/VSD; canine radiculomegaly & other dental anomalies |
| Osteoporosis-Pseudoglioma Syndrome (OPPG) | 259770 | AR | 11q13.4 (Low density lipoprotein receptor-related protein 5) | Osteogenesis imperfecta; retinal dysplasia, PHPV; corneal opacity; secondary glaucoma; pseudoglioma; hypotonia; ligamentous laxity |
| Steinfeld Syndrome | 184705 | AD | Unknown | Holoprosencephaly; ulnar/radial hypoplasia w/ phocomelia; renal dysplasia; CL/CP (median); Tetralogy of Fallot; vertebral/rib anomalies |
| Solitary median maxillary central incisor (SMM1) | 147250 | AD | 7q36 (SHH, sonic hedgehog) | Single central incisor; holoprosencephaly; short stature w/ growth hormone def.; coloboma; ectodermal dysplasia |
| Walker-Warburg syndrome | 236670 | AR | 9q34.1 (protein Omannosyltrans-ferase) | Hydrocephalus; agyria; retinal dysplasia/congenital retinal detachment; encephalocele; AC abnormalities; cataract; glaucoma; coloboma; Peters' anomaly; PHPV; CL/CP; imperforate anus; GU anomalies; lissencephaly; seizures |
| Warburg Micro syndrome 1 (WARBM1) | 600118 | AR? | Unknown | Microcornea; cataract; abnormal ERG; optic atrophy; microcephaly; MR; hypogenitalism; |
Sp, sporadic; XLD, X-linked dominant; AR, autosomal-recessive; AD, autosomal-dominant; CL, cleft lip; CP, cleft palate; ASD, atrial-septal defect; VSD, ventriculo-septal defect; MR, mental retardation; AC, anterior chamber; PHPV, persistent hyperplastic primary vitreous; GU, genitourinary.
Numerous chromosomal abnormalities have been associated with microphthalmia (Table 5).63 Warburg and Friedrich provided a comprehensive review of these disorders in 1987.64 Presumably, any chromosomal disorder that significantly affects telencephalon or cranial neural crest development could result in microphthalmia.
TABLE 5. Chromosomal Abnormalities Associated With Microphthalmia
| Chromosomal Abnormality | Other Features |
| Duplication 3q syndrome (3q21->ter dup) | Mental & growth deficiency; broad nasal root; hypertrichosis; craniosynostosis; anteverted nares; cardiac defects; chest deformities; genital abnormalities; umbilical hernia |
| 4p- (Wolf-Hirschhorn syndrome) | Growth deficiency; microcephaly, ocular hypertelorism with broad or beaked nose; Peters anomaly; cranial asymmetry; low-set, simple ears with preauricular dimple; MR; seizures; CL and/or CP; anterior segment dysgenesis (occasional) |
| Duplication 4p syndrome | MR; seizures; growth deficiency; obesity; microcephaly; characteristic faces; genital abnormalities; kyphoscoliosis |
| Trisomy 9 mosaic syndrome | Joint contractures; congenital heart defects; low-set, malformed ears; prenatal growth deficiency; MR; sloping forehead; micrognathia; fleshy nasal tip with slit-like nostrils; narrow chest; kyphoscoliosis; hypoplastic toes |
| Duplication 10q syndrome | Ptosis, short palpebral fissures, camptodactyly, MR, prenatal growth deficiency, microcephaly, heart & renal malformations |
| 13q-, 13 ring | Microcephaly with MR (brain abnormalities; high nasal bridge; bilateral retinoblastoma; thumb hypoplasia; cardiac defects; hypospadias, cryptorchidism |
| Trisomy 13 (Patau syndrome) | Holoprosencephaly; moderate microcephaly; sloping forehead; coloboma; retinal dysplasia; CL and/or CP; posterior scalp defects; abnormal external ears w/ deafness; polydactyly; narrow hyperconvex fingernails; cardiac defects; genital abnormalities; single palmar crease; prominent heel |
| 18q- | Midface hypoplasia, prominent antihelix, whorl digital pattern, small stature, MR, hypotonia, nystagmus, conductive deafness, microcephaly, midface hypoplasia, vertical talus, genital abnormalities |
| Trisomy 18 (Edward's syndrome) | Clenched hand; short sternum; low-arch dermatoglyphics; polyhydramnios; single umbilical artery; small placenta; feeble fetal activity; MR; hypertonicity; hypoplasia of skeletal muscle, subcutaneous, adipose tissue; prominent occiput; low-set malformed auricles; micrognathia; absent distal finger creases; cardiac defects |
| Triploidy syndrome | Large placenta w/ hydatidiform changes, growth deficiency, syndactyly of 3rd/4th fingers, congenital heart defects, brain anomalies/holoprosencephaly |
Abnormalities associated with microphthalmia. Only the more common chromosomal abnormalities are listed. The reader is referred to Warburg and Friedrich (1987) for a more complete discussion.64 MR, mental retardation; CL, cleft lip; CP, cleft palate.
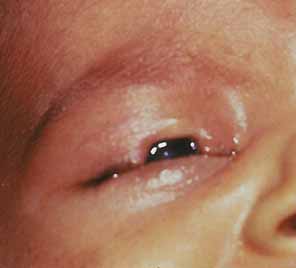
Complex microphthalmia is often associated with uveal colobomas. In the most common use of the term, “coloboma” refers to a defect in the iris, retina/uvea, and/or optic nerve as a result of faulty closure of the embryonic fissure at the 7- to 20-mm stage of development. Colobomas tend to be inferonasal, which is the normal position of the embryonic fissure (Fig. 1). The term “coloboma” has also been applied to other anomalies of the optic nerve, the macula, and the uveal tract that are not related to faulty closure of the embryonic fissure. This loose use of terminology creates unnecessary confusion when describing a syndrome. Hornby et al. found in their series that 72 of 185 eyes with coloboma also were microphthalmic and 71 of those 72 had microcornea.65 Thus, not every patient with microphthalmia has coloboma and not every patient with coloboma has microphthalmia. Microphthalmia has been postulated to arise from faulty development of the secondary vitreous and, hence, not enough pressure needed for prenatal eye growth.66
|
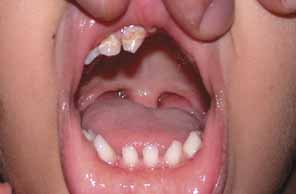
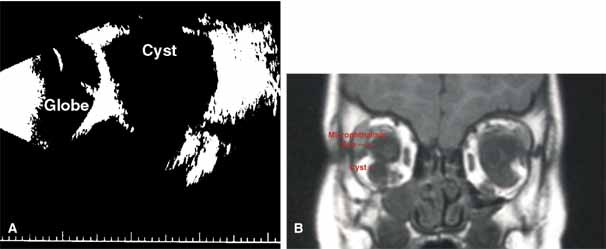
“Microphthalmia with cyst” refers to cases of colobomatous microphthalmia with a posterior eye wall defect through which a cyst lined with neuroectodermally derived tissue protrudes into the orbit. These cysts may be small and only detected by imaging (Fig. 2); sometimes, they may be large enough to cause progressive proptosis. Most cases are isolated, but familial occurrences have been reported.67–69 Inheritance is likely autosomal recessive. Depending on the size of the cyst, its appearance, and the visual potential of the microphthalmic eye, these cysts may be managed by observation, excision with or without enucleation, and/or aspiration.70 Aspirated cysts tend to reaccumulate fluid. Socket expansion improves cosmesis.
|
Complex microphthalmia can also be seen as part of Peters anomaly and persistent hyperplastic primary vitreous (PHPV). These disorders are covered in the anterior segment and retina/vitreous sections of this chapter.
Not all cases of complex microphthalmia are genetically determined. Intrauterine infection and maternal exposure to alcohol or other teratogens can also cause complex microphthalmia.
Percin et al. described mutations in the homeobox gene CHX10 (14q24.3) in two families with autosomal recessive, nonsyndromic microphthalmia, iris abnormalities, and coloboma.71 Like other homeobox proteins, CHX10 is a transcription factor that binds to specific DNA sequences in the regulatory regions of other genes and affects their transcription during development. Chx10 mutations were previously found in the ocular retardation mouse model, which is characterized by microphthalmia, a thin hypocellular retina, and optic nerve aplasia.72
One form of autosomal recessive complex microphthalmia was found by Bessant et al. to map to 14q32.73 Phenotypic features included sclerocornea (8/8 patients) with secondary corneal vascularization (6/8 patients) and staphyloma formation (3/8 patients), elevated intraocular pressure (5/8 patients), and short axial length (5/5 measured).74 These authors ruled out CHX10 and OTX2 as candidate genes in this pedigree.
Lehman et al. reported a large Mexican American pedigree with isolated X-linked recessive colobomatous microphthalmia that maps to the proximal p or q arm of the X chromosome.75
Microphthalmia can usually be diagnosed by inspection and/or palpation of the eye through the eyelids. The presence of a bulge in the lower lid usually indicates microphthalmos with cyst (Fig. 3). Axial length measurements may aid in the diagnosis of simple microphthalmia/nanophthalmos. Refraction and amblyopia treatment may be helpful in cases in which the macula is well developed. In cases in which no useful vision is present, scleral shells may improved cosmesis. Oculoplastics procedures to enlarge the palpebral fissures and the socket are sometimes helpful.
|
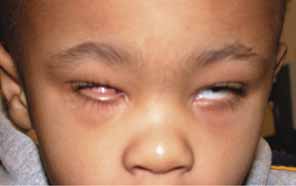
CONGENITAL CYSTIC EYE
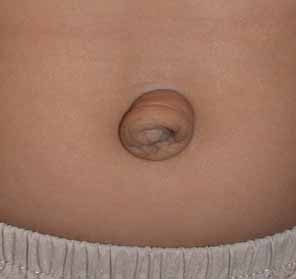
Congenital cystic eye results from failure of the optic vesicle to invaginate, producing a cystic structure that may contain elements of dysplastic retina or lens material.76 These cysts do not contain an epithelial lining, may not be evident at birth, and may grow with time. Approximately 30 cases have been described in the literature. Most cases are unilateral, but bilateral developmental eye anomalies have been described.77–80 Orbital imaging can aid in the diagnosis and preoperative planning.81
Oculocerebrocutaneous syndrome (Delleman syndrome, OMIM 164180)82 is a sporadic condition associated with orbital cysts, focal dermal hypoplasia, periorbital skin appendages, and cerebral malformations. Anophthalmia may also occur in patients with this syndrome.
PERSISTENT HYPERPLASIA PRIMARY VITREOUS (PHPV) AND PERSISTENCE OF FETAL VASCULATURE (PFV)
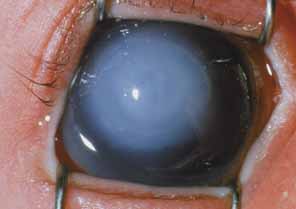
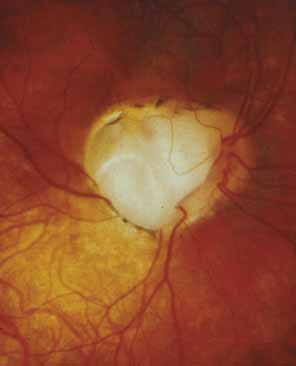
Persistent hyperplastic primary vitreous (PHPV) or persistence of fetal vasculature (PFV) is a complex malformation of the eye characterized by the presence of remnants of the hyaloid and of the tunica vasculosa lentis systems of blood vessels, together with proliferation of fibrovascular tissue behind the lens, and variable degrees of posterior pole and/or anterior retinal dysplasia.83,84 The eye in PHPV is generally microphthalmic but can be enlarged with accompanying myopia.85 Although PHPV primarily affects the posterior part of the eye, the ciliary body, iris, and lens are involved to a variable extent. In the typical case, ciliary processes are elongated and converge to a plaque of retrolental fibrovascular tissue that may involve the lens capsule proper (Fig. 4). The lens may be completely clear or may be cataractous. The fibrovascular plaque may adhere to the posterior lens capsule and vessels can invade the lens—a finding pathognomonic of PHPV. The retrolental membrane may contain adipose tissue, cartilage, and smooth muscle tissue. This is thought to be the result of metaplastic changes in tissues of mesenchymal origin. In a series of 47 eyes, Font et al. found adipose tissue in 10 and cartilage in one.86 The peripheral retina may also be drawn anteriorly into the retrolental membrane. In what has been referred to as “posterior PHPV,” the retina around the disc is pulled up into the posterior vitreous and is thrown into folds that may involve the macular and lead to poor vision. The retina is generally normal in structure, but dysplastic changes have been described. Vitreoretinal traction may lead to retinal breaks and detachment. The fellow eye in PHPV is usually normal but may have a Mittendorf dot or, rarely, one of other abnormalities.
|
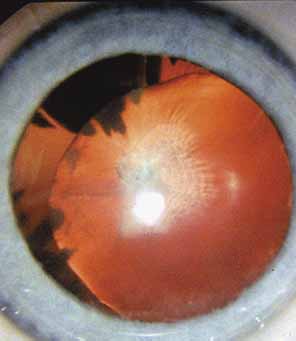
PHPV is unilateral in approximately 90% of cases. The globe varies in size from normal to moderately decreased, being slightly smaller than normal in the majority of cases. The cornea is clear. The anterior chamber is shallow in smaller eyes because of anterior displacement of the iris/lens diaphragm; this predisposes patients with PHPV to angle-closure glaucoma, which usually develops later in life. Lens extraction prevents secondary angle-closure glaucoma. The iris may be normal but frequently shows small notches at the pupillary margin, where iridohyaloidal vessels were located in the developing eye and failed to regress as the iris matures and the tunica vasculosa lentis resorbs. Patent iridohyaloidal vessels may be observed to course over the anterior iris surface, the pupillary margin, and the posterior iris surface to anastomose with vessels in the retrolental membrane. These iridohyaloidal vessels are characteristic of PHPV.
Clinically, patients with PHPV present with a small eye since birth, a white pupillary reflex from a cataract or retrolental membrane, and/or strabismus because of poor vision. Retinoblastoma should be ruled out using clinical examination, ultrasonography, and computed tomography.87 There is no intraocular calcification in PHPV.88 Retinoblastoma generally occurs in normal-size eyes except in patients with 13q deletions and microphthalmia. PHPV and retinoblastoma have been rarely reported together in a microphthalmic eye.89
PHPV and its variants are not very rare. They are a common cause of unilateral congenital cataracts. PHPV has also been reported in patients with recessive oculodento-osseous dysplasia,47 in one patient with protein C deficiency,90 in the oculopalatocerebral syndrome, and in two generations of one family with Rieger anomaly.91
PHPV may be the result of defective formation of the secondary vitreous, which is derived from the inner retinal cells starting in the ninth week of gestation. The secondary vitreous compresses the regressing primary vitreous, which is probably derived from mesenchyme and contains the hyaloid system of blood vessels that anastomose with the tunica vasculosa lentis anteriorly. The globe remains small because its growth depends partly on the expansion of the secondary vitreous.92 The cause of PHPV has not been identified yet. Familial occurrences of PHPV have been reported in dizygotic twins, in two brothers, and in a mother and son.93
Visual prognosis in PHPV is generally guarded. Early cataract surgery may result in relatively good visual results in selected patients.94,95 Eyes with anterior PHPV have a better vision potential than those with posterior pole dysplasia. An anterior approach to lens extraction and retrolental membrane removal is recommended in mild anterior cases. A combined lensectomy-vitrectomy using a pars-plana surgical approach should be considered in patients with significant mid-vitreal or posterior vitreal components to the PHPV. Tractional and rhegmatogenous retinal detachments have been reported in patients with PHPV and are treated surgically as needed.
SYNOPHTHALMIA/CYCLOPIA
Cyclopia refers to a single, midline eye; synophthalmia refers to an apparent midline fusion of two eyes. These conditions are points along a phenotypic continuum. Clinical findings vary but include a proboscis above the eye(s) and forebrain malformations. Many fetuses with cyclopia or synophthalmia have chromosomal abnormalities and are spontaneously aborted.96
Formation of a cyclopic or synophalmic globe may occur by one of two embryologic mechanism.97 At the trilaminar embryo stage, a field of bilobed, midline ectodermal tissue is “fated” to become the eyes. Failure of this bilobed area to separate completely into two fields could result in synophthalmia or cyclopia. Alternatively, previously separated globes could fuse as a result of faulty midline development.
The latter mechanism is likely responsible for the cyclopia and synophthalmia seen in holoprosencephaly. Holoprosencephaly is a genetically and phenotypically heterogeneous group of disorders that manifest with midline abnormalities.98,99 Classically, this includes complete (alobar) or partial (semilobar) “fusion” of the normal, bilateral cerebral hemispheres into a single hemisphere. Mutations in several genes important in forebrain development have been identified in families with holoprosencephaly, including sonic hedgehog (SHH, 7q36), ZIC2 (13q32), SIX3 (2p21), and transforming growth factor beta-induced factor (TGIF, 18p11.3).100–103
ANIRIDIA
Classic aniridia is a sporadic or autosomal dominant, bilateral, pan-ocular condition caused by haploinsufficiency of the PAX6 homeobox gene on 11p13.104,105 Contrary to the connotation of “aniridia,” most aniridic patients have at least some rudimentary iris tissue. “Aniridia” is also used to describe absence of iris tissue in some syndromes that are possibly unrelated to PAX6 haploinsufficiency (see later).
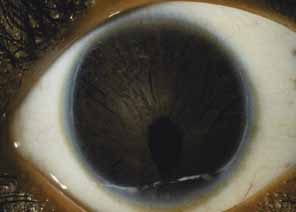
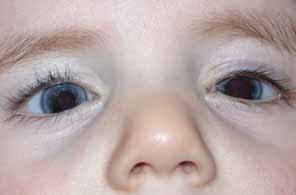
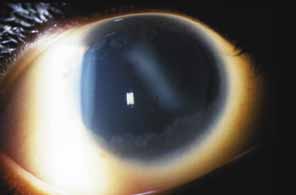
Aniridia affects multiple tissues in the eye and has a broad range of clinical presentations. Iris abnormalities range from almost complete absence to radial iris clefts to iris hypoplasia (Fig. 5).106–109 Foveal hypoplasia is characteristic, but of varying severity, which include optic nerve hypoplasia, corneal pannus, and/or a superficial corneal dystrophy (Fig. 6), microcornea, Peters anomaly, congenital cataracts, ectopia lentis, and glaucoma.110,111–114 Visual potential varies with the severity of the aniridic phenotype. Vision-limiting factors include optic nerve and foveal hypoplasia, cataracts, and—occasionally—the keratopathy that arises in older patients with aniridia. The keratopathy/corneal pannus is presumably caused by insufficient/absent limbal stem cells. Nystagmus develops in aniridic patients, presumably caused by congenital poor visual acuity. Iris vascular anomalies and leakage have been diagnosed on fluorescein angiography of the anterior segment.109
|
|
Shaw et al. estimated the prevalence of aniridia in the lower peninsula of Michigan in 1960 to be approximately 1 in 64,000.115 Approximately two-thirds of patients have at least one other affected family member; the remaining one-third are sporadic.
Pathologic studies of aniridic eyes are sometimes complicated by the presence of advanced glaucoma and/or a history of multiple surgical interventions. In 1983, Margo described pathologic features of seven eyes from seven children 6 months to 14 years of age.116 None of these eyes had advanced glaucoma or previous surgery. Congenital anomalies included iris and ciliary body hypoplasia, attenuation of Bowman membrane, and (in two eyes from children with 11p deletions) anomalous anterior chamber angles. Other abnormalities included corneal pannus, cataracts, and peripheral anterior synechiae. Resolution was insufficient to comment on retinal or optic nerve anomalies.
Elsas et al. classified aniridia into four types based on clinical criteria.107 Type I aniridia—what we call classic aniridia—is autosomal-dominant and is characterized by near-absence of iris tissue, foveal hypoplasia, nystagmus, corneal pannus, optic nerve hypoplasia, secondary glaucoma, and (sometimes) dislocation of the lens. Type II aniridia patients have varying iris abnormalities, no nystagmus, and relatively preserved visual acuity. Patients with type III aniridia have classic aniridia and mental retardation. Type IV aniridia patients also have genitourinary anomalies, an increased risk of Wilmss tumor, and mental retardation (WAGR syndrome, discussed later). This system was devised before our detailed knowledge of the cytogenetic and molecular causes of aniridia. Therefore, it does not take into account more recent findings of the phenotypic, cytogenetic, and mutational heterogeneity of this disorder.
Grant and Walton noted progressive angle changes in aniridic patients.117 Although the trabecular meshwork of 25 aniridic patients who did not develop glaucoma appeared normal on gonioscopy, the angles of 31 aniridic patients with glaucoma showed progressive obstruction of the trabecular meshwork by iris tissue. These authors recommended prophylactic goniotomy in aniridic patients who show progressive angle changes before glaucoma develops. This recommendation has not been studied in a controlled fashion.
Cytogenetic studies have played a pivotal role in understanding the molecular basis of aniridia. WAGR syndrome (OMIM 194072)—an acronym for Wilms tumor, aniridia, genitourinary abnormalities, and mental retardation—is associated with deletions of chromosome 11p13 (reviewed by Mannens et al.).118 By comparing the phenotype of patients with the size of the deletion, as determined using both cytogenetic and molecular markers, the critical region for aniridia and for Wilms tumor were identified. Other cytogenetic abnormalities such as paracentric inversions, balanced translocation, and unbalanced translocation involving 11p13 have been reported in subjects with aniridia, both with or without systemic findings.119,120 Not all chromosomal aberrations directly disrupt the PAX6 gene, suggesting that local chromatin structure or cis-acting elements are important for PAX6 expression.121 Crolla and van Heyningen have shown that some patients with deletion of both the Wilms tumor gene (WT1) and PAX6 may not be cytogenetically visible and can only be detected with fluorescent in-situ hybridization (FISH).120
Because the PAX6 gene and the Wilms tumor gene (WT1) are on the same 700-kilobase region of 11p13, chromosomal changes that cause aniridia may also affect the WT1 locus. In general, the increased risk of Wilms tumor occurs in sporadic aniridia cases. The exception to this rule is families in which an affected parent has a deletion of WT1 and passes his/her chromosomal abnormality onto their child. In these cases, autosomal-dominant inheritance is present. Not everyone with a WT1 deletion develops Wilms tumor. Gronskov et al. found two out of five subjects with WT1 deletions that developed Wilms tumor.122 Muto et al. found 13 (45%) of 29 patients with deletions in the Wilms tumor critical region developed Wilms tumor.123 Therefore, cytogenetic studies focused on 11p13 are reasonable in both sporadic aniridia and familial aniridia, when the number of affected individuals in the family is small.
In 1989 Mannens et al. mapped the aniridia locus to markers on 11p13.124 Further analysis of an aniridia pedigree originally mapping to chromosome 2 (AN1 locus)125 showed that the true linkage in this family was also to 11p13 (the AN2 locus.) In 1991, Ton et al. discovered that a homeobox transcription factor gene, now know as PAX6, was either totally or partially deleted in two patients with aniridia.104 Since then, numerous reports of mutations in PAX6 have been reported in patients with aniridia.105 A detailed database of PAX6 mutations can be found online at http://pax6.hgu.mrc.ac.uk/.
The PAX6 gene covers approximately 25 kb of DNA, consists of 14 exons, and codes for a 422-amino acid transcription factor.105 This 422-amino acid protein contains two distinct motifs that bind to a range of DNA target sequences—a paired domain and a homeodomain.126,127 (The terms “paired domain” and “homeodomain” are applied to these portions of the protein because they strongly resemble similarly named genes important in body segmentation in Drosophila). DNA binding is modified by alternative splicing of one portion of the paired domain, which affects the target sequence of DNA recognized. Translation begins at exon 4 and one exon (5a) is alternatively spliced. PAX6 protein is highly conserved across vertebrates. PAX6 is expressed in all layers of the neuroectoderm that forms the developing eye, as well as in the surface ectodermal cells that become the lens.128
Nearly all patients with classic aniridia have mutations predicted to cause loss of function of one PAX6 allele (see http://pax6.hgu.mrc.ac.uk/). In general, the ocular phenotype of these patients is indistinguishable from patients with complete deletion of the gene, implying that haploinsufficiency is the mechanism of disease. Patients with PAX6 missense mutations can have aniridia or a variety of other ocular phenotypes—corneal pannus/keratopathy, isolated foveal hypoplasia, congenital cataracts, Peters anomaly, anterior segment dysgenesis, uveal ectropion, optic nerve hypoplasia, optic nerve coloboma/morning glory discs, and persistent hyperplastic primary vitreous.114,129–134,135 For example, Kivlin et al. reported on what appeared to be a new, autosomal-dominant keratopathy (ADK) associated with ocular irritation and photophobia, and anterior stromal vascularization and inflammation.136 Pearce et al. suggested that ADK was an aniridia variant based on the presence of iris abnormalities and macular hypoplasia in a four-generation family with ADK.110 Mirzayans et al. subsequently found splice-site mutations in PAX6 in a family with ADK.137
The precise mechanism behind this phenotypic variability is unclear. Presumably, other gene products and environmental factors regulate or interact with PAX6 during ocular development; variations/abnormalities in these proteins lead to the variability in the aniridia phenotype. A few putative target genes activated by PAX6 have been identified, including lens crystallins, cell adhesion molecules like L1 and NCAM, glucagon, and insulin.138–142
Heterozygous small eye (Sey) mice have disrupted expression of one allele of Pax6 and are considered a model of human aniridia.143–145 The phenotype is variable, as in humans, and includes iris abnormalities, cataract, Peters anomaly, microphthalmia, and keratopathy.146
Treatment of aniridia centers around the treatment of its individual ophthalmic manifestations. As always, aggressive treatment of amblyopia in children and optimal refraction in all patients should be pursued. Cataracts should be removed if visually significant during the amylogenic period or later in life if visual deterioration appears to be linked to cataract progression. Some success has been reported with sulcus-based intraocular lenses and with black diaphragm aniridia lenses in these patients.147,148 Glaucoma is common and intraocular pressure should always be checked. It can be quite difficult to manage and often requires surgery. Numerous methods, including trabeculotomy, seton implantation, and ciliary body ablation, have had some success.149–152 As previously mentioned, some advocate prophylactic goniotomy in patients with progressive angle changes.153 No clear consensus exists on how to approach this difficult clinical problem.
The corneal pannus and ocular surface problems associated with aniridia can also cause visual deterioration. Penetrating keratoplasty is not usually successful because it does not treat the presumed, underlying limbal stem cell deficiency.154,155 Holland et al. have reported success with keratolimbal allografts in patients with aniridia.156 In their study, 74% of eyes had an improved ocular surface, mean visual acuity increased from 20/1000 to 20/165, and 70% of those undergoing secondary penetrating keratoplasty had successful grafts. However, continued use of immunosuppressive drugs appears to be required for optimal results.
Syndromes With Associated Aniridia
Warburg et al. described a girl with aniridia, mental retardation, gonadal streaks, gonadoblastoma, hearing loss, kidney malrotation, and dysmorphic facies that had an interstitial deletion between bands 11 and 13 on 11p.157 As discussed previously, the constellation of Wilms tumor, aniridia, genitourinary anomalies, and mental retardation (WAGR) is caused by deletions in this region of chromosome 11. Identification of patients with such cytogenetic abnormalities was critical in the isolation of PAX6 as the gene for human aniridia.
Pupil abnormalities reminiscent of aniridia are also seen with cerebellar ataxia and mental retardation in a condition often referred to as Gillespie syndrome. (The original patients described by Gillespie, however, have some features such as congenital cataracts that are not part of what is now referred to as “Gillespie syndrome”).158 The pupil in these patients appears constantly dilated and often has iris strands adherent to the lens. Unlike classic aniridia, patients with Gillespie syndrome generally do not have cataracts, corneal abnormalities, or foveal hypoplasia. Visual acuity has not been consistently recorded in the literature, but may be reduced.159 The nystagmus seen in these patients appears to be more related to cerebellar and other CNS pathology than to sensory issues. Although inheritance is generally thought to be autosomal recessive, Nelson et al.. have questioned whether some autosomal dominant cases might exist.159 This same group has found extensive white matter and cerebral abnormalities in one child with Gillespie syndrome, suggesting that the brain pathology may extend beyond the cerebellum. Glaser et al. did not find mutations in the PAX6 gene and the phenotype did not segregate with genetic markers on 11p in families with Gillespie syndrome.160
Mirkinson and Mirkinson described a family with aniridia and hypoplastic/aplastic patellae.161 The proband's grandmother had glaucoma, but it is unclear if this was of childhood onset. No other stigmata of classic, PAX6-associated aniridia are mentioned. No mutations have been reported in this pedigree.
Hamming et al. reported a mother and her two children with variable iris abnormalities, ptosis, nystagmus, foveal hypoplasia, corneal pannus, and cataracts.162 The mother had unspecified cardiac anomalies, recurrent miscarriages, and alopecia. One of the children was moderately mentally retarded and both children were obese. These authors suggest that this family has a rare, autosomal-dominant aniridia variant. Although chromosomal studies were reportedly normal, PAX6 mutation testing was not performed.
Although Levin et al. reported a child with aniridia, congenital glaucoma, hydrocephalus, and a ring chromosome 6 {46 XY, r(6) (p24→q26)}, the “aniridic” iris abnormalities were unilateral.163 The contralateral eye had findings more consistent with Axenfeld-Rieger syndrome. Ring chromosomes are formed when the long (q) and short (p) arms of a chromosome fuse together. Such joining is invariably associated with the loss of genetic material from one or both chromosomal arms. In this case, the patient was missing genetic material distal to p24 on the short arm and to q26 on the long arm of chromosome 6. This patient's ocular findings might be explained by a deletion of the FOXC1 gene on 6p25 (see discussion of Axenfeld-Rieger syndrome).
Walker and Dyson report on an association of classic aniridia with mental retardation.164 Although most patients with classic aniridia are intellectually normal, PAX6 is expressed in the developing brain and it is conceivable that in some individuals it may contribute to a mental handicap.