Table 1. Primary Immunodeficiency Disorders
- Combined immunodeficiencies
- Severe combined immunodeficiency
- T-lymphocyte-deficient variant
- T- and B-lymphocyte-deficient variant
- T-lymphocyte-deficient variant
- X-linked hyper-immunoglobulin M syndrome
- Purine nucleoside phosphorylase deficiency
- Major histocompatibility complex class II deficiency
- CD3 deficiency
- Zap-70 deficiency
- TAP-2 deficiency
- Severe combined immunodeficiency
- Predominantly antibody deficiencies
- X-linked agammaglobulinemia
- Non-X-linked hyper-IgM syndrome
- Immunoglobulin heavy-chain deletions
- Autosomal recessive K-chain deficiency
- Selective immunoglobulin subclass deficiency
- Antibody deficiency with normal immunoglobulin
- Common variable immunodeficiency
- IgA deficiency
- Transient hypogammaglobulinemia of infancy
- Autosomal recessive agammaglobulinemia
- X-linked agammaglobulinemia
- Predominantly T-lymphocyte deficiencies
- CD4 deficiency
- CD7 deficiency
- Interleukin-2 deficiency
- Multiple cytokine defect
- Signal transduction defect
- Calcium flux defect
- CD4 deficiency
- Other well-defined syndromes
- Wiskott-Aldrich syndrome
- Ataxia-telangiectasia
- DiGeorge's anomaly
- Wiskott-Aldrich syndrome
- Complement deficiencies*
- Congenital defects of phagocytic number and/or function
- Severe congenital neutropenia
- Cyclic neutropenia
- Leukocyte adhesion defect
- Chédiak-Higashi syndrome
- Specific granule deficiency
- Schwamann's syndrome
- Chronic granulomatous disease
- Neutrophil G6PD deficiency
- Myeloperoxidase deficiency
- Interferon-γ deficiency
- Severe congenital neutropenia
*Defects of almost all components have been described.
(Adapted from Rosen FS, Wedgewood RJ, Eibl M et al. Primary immunodeficiency diseases. Report of a WHO scientific group. Clin Exp Immunol 1997;109[Suppl]:1.)
Our knowledge of the abnormal genes and altered biochemical pathways that result in primary immunodeficiency diseases has increased dramatically in the last decade. More than 60 genetic loci have been identified that are associated with specific defects in the immune system.10 Abnormalities in the production of cell surface receptor molecules, cytoskeletal proteins, enzymes involved in purine and oxidative metabolism, immunoglobulins, complement components, and cytokines have been described; in addition, defects in intracellular signaling pathways and T-lymphocyte cell receptors, as well as defective rearrangements of immunoglobulin genes, have been identified. Understanding the genetics and pathophysiology of these abnormal states has provided new insights into the functioning of the immune system in health and disease.
Distinct and disparate immunologic diseases such as X-linked agammaglobulinemia, C3 deficiency, and neutropenia can produce similar clinical features, even though they are caused by unrelated pathogenic mechanisms affecting different components of the immune system. With selective defects of immune function, other intact immune mechanisms may partially compensate for the deficiency and further blur any clinical distinction between disorders. A similar phenomenon has been found in gene knock-out mice, strains that lack very specific effectors of immune function; they reveal redundancies in the immune system that, although confusing in research models, can be critical for maintaining function. Some diseases, however, do have unique clinical features that aid in establishing a diagnosis.
It has been estimated that in the general population approximately 2500 persons per million (0.25%) have primary immunodeficiencies, most of whom are male.11 Selective immunoglobulin A (IgA) deficiency is believed to be the most common primary immunodeficiency, with a reported prevalence of 1:700 in whites.2 It is estimated that agammaglobulinemia occurs in 1 of 50,000 live births and severe combined immunodeficiency in 1 of 100,000 to 1 of 500,000 live births.3
LYMPHOCYTE DISORDERS
Predominantly Antibody Defects
Defects of humoral immunity may involve all or only selected types of antibodies. Antibody deficiency alters several protective immunologic mechanisms, thus increasing the frequency, chronicity, and severity of infections caused by various bacterial, viral, and protozoan pathogens. Lack of secretory antibodies, which usually accompanies a deficiency of serum antibodies, increases host susceptibility to attachment of bacteria, viruses, and parasites to mucosal surfaces.4 Gastrointestinal (GI) and respiratory tract infections therefore predominate, and the ocular surface may be at increased risk. Systemic seeding of pathogens and violation of the blood-brain barrier may occur more readily because levels of antibodies that function in neutralization, cytotoxicity, or antibody-dependent cellular cytotoxicity reactions are low. Failure to form antibody-antigen complexes limits the actions of the innate, nonspecific immune defense mechanisms normally called into play, such as activation of the complement system with subsequent chemotaxis of phagocytic cells.
Bacterial pathogens commonly associated with antibody deficiencies include many encapsulated organisms, such as Streptococcus pneumoniae, Haemophilus influenzae, Staphylococcus aureus, and Neisseria meningitidis. Common viral agents include enteroviruses and rotaviruses. Giardia lamblia and Cryptosporidium species are considered the most common protozoa that cause disease in these patients. Infections with Pneumocystis carinii, (previously classified as a protozoan parasite but now considered to be more closely related to the fungi12), as well as infections with cryptosporidium, occur in patients with X-linked hyper-IgM syndrome.13,14
In addition to recurrent pneumonia and chronic diarrhea, infectious disorders associated with antibody deficiencies include sinusitis, otitis, viral hepatitis, paralytic poliomyelitis, arthritis related to Mycoplasma sp., cystitis, and urethritis. Meningitis and viral encephalitis are the most important complications. Ocular disorders usually involve the eyelid, conjunctiva, or cornea. In most cases, they are attributed to recurrent bacterial infection.
Sjögren's syndrome and other autoimmune diseases have been associated with immunoglobulin deficiencies,15–17 most commonly with IgA deficiency and common variable immunodeficiency. Those with primary Sjögren's syndrome may be deficient in the IgG2 subclass as well.16 Infiltration of the salivary glands by lymphocytes is observed. It is possible that xerophthalmia will be more commonly recognized as experience with immunodeficient patients increases.
Described subsequently are several immunodeficiency disorders for which there have been specific references to associated ophthalmic disorders. There is no evidence, however, that there are unique ocular changes in any of the specific antibody deficiency syndromes.
X-LINKED AGAMMAGLOBULINEMIA.
X-linked agammaglobulinemia (Bruton's agammaglobulinemia) is associated with a deficiency of a tyrosine kinase that is required for B-lymphocyte maturation; the gene for this enzyme is found on the X-chromosome. Patients with X-linked agammaglobulinemia have repeated bouts of pyogenic infections. Keratoconjunctivitis has been reported in these patients,18 but in most cases, the eyes remain unaffected.19,20 Eczema is common in this condition,21 and involvement of the eyelids might be expected.
SELECTIVE DEFICIENCY OF IMMUNOGLOBULIN ISOTYPES.
Some patients with IgG4 deficiency have multiple hordeola and chalazia, marginal corneal infiltrates, and vascularization of the limbus.22 Other cases, however, have no reported ocular findings.23
Little information is available about ophthalmic involvement with other selective antibody disorders. Selective IgM deficiency results in severe eczema, verrucae vulgaris, and recurrent bacterial infections of the skin.21 Periorbital cellulitis has been reported with this condition.3 Patients who have immunodeficiency with hyper-IgM develop severe verrucae vulgaris,21 which could involve the eyelids.
SELECTIVE IGA DEFICIENCY.
IgA deficiency is found in 1:700 whites but in only 1:18,500 Japanese.2 Ocular infection with IgA deficiency is believed to be rare.9,24 Ocular disease is more common when other immunoglobulin classes are also grossly affected.18 In cases in which low serum levels of IgA do not reflect a deficit in secretory IgA, the ocular surface may be at no increased risk for infection. It may be dangerous to attempt IgA replacement; patients with IgA deficiency may have anaphylactic reactions to IgA-containing fluids.25 Even if IgA replacement in blood were possible, it would not protect the ocular surface in most cases; IgA cannot be transported into tears without a secretory piece.
Hemorrhagic retinitis was associated with IgA deficiency in one patient, but it may actually have been the result of systemic lupus erythematosus in that situation.26 Retinal vasculitis has been described in patients with common variable immunodeficiency, which may be a related syndrome, because it is associated with low levels of serum IgA in some people.27 IgA levels in those patients were not reported, however.
An infant with an unusual phacomatosis that included findings of oculocutaneous pigmentation, neurologic abnormalities, and cutaneous vascular abnormalities also had an IgA deficiency.28 He had unilateral glaucoma and abnormal intraocular vessels, described as being similar to those found in Sturge-Weber syndrome.
COMMON VARIABLE IMMUNODEFICIENCY.
Common variable immunodeficiency is associated with decreased levels of serum IgA and IgG (and in some cases IgM). Approximately 25%; of cases are familial. The genetic defect is not known. There is an equal gender distribution. There are two peaks of disease onset, at ages 1 to 5 years and 16 to 20 years. Patients may have been immunologically normal before onset of IgA deficiency. Patients with IgA deficiency may progress to common variable immunodeficiency. This disorder resembles X-linked hypogammaglobulinemia clinically; the spectrum of associated disorders is similar, although infections are usually less severe and have later onset.3 In some cases, medications (e.g., sulfasalazine, hydantoin, and carbamazepine) have been implicated as causes.29
Franklin and associates18 reported on five patients, four of whom had conjunctivitis or keratoconjunctivitis. H. influenzae was isolated from three of these patients, one of whom also was found to be infected with S. aureus; Staphylococcus epidermidis was isolated from the fourth patient. Ocular findings are believed to be rare in this syndrome, however. Among approximately 500 cases in the United Kingdom, one patient was reported to have acute unilateral uveitis30 and another acute annular detachment of the choroid and retina with panuveitis (William J. Dinning, unpublished data). These findings may have been incidental; they could not be attributed specifically to the immunodeficiency. A patient with common variable immunodeficiency has been reported with severe corneal thinning that was thought to be sterile in both eyes.31 The problem progressed to corneal perforation in his right eye and a secondary H. influenzae endophthalmitis developed, with loss of the eye. The fellow eye was treated with a lamellar corneal graft, and the patient had 20/25 vision in that eye until his death 2 years later.
Autoimmune diseases, including Sjögren's syndrome, systemic lupus erythematosus, and rheumatoid arthritis have been associated with IgA deficiency and common variable immunodeficiency. Retinal vasculitis, presumed to be autoimmune in origin, has been reported in three patients with common variable immunodeficiency.27 Associated findings included macular edema, vitreitis, optic disc edema, and retinal neovascularization. No evidence of infection was found. Therapy with periocular injections of corticosteroids, oral prednisone, and cyclosporine appeared to be responsible for maintaining 20/40 or better vision in two patients, but visual acuity dropped to 5/200 in both eyes of the third patient.
Combined Immunodeficiencies
SEVERE COMBINED IMMUNODEFICIENCY DISEASE (SCID).
The combined immunodeficiencies are a group of disorders in which both T- and B- lymphocyte function is abnormal. The abnormalities that have been found appear to be primarily in T lymphocytes, highlighting the importance of these cells for normal B-lymphocyte function, as well as in cell-mediated immunity. Defects in the common γ chain of several cytokine receptors, in intracellular signal transduction, in Class II major histocompatibility complex (MHC) expression, in enzymes involved in purine metabolism, and in T-lymphocyte receptor gene rearrangement have all been described in patients with combined T- and B-lymphocyte immunodeficiencies.10
SCID is a designation that refers to potentially fatal, profound defects in both cellular and humoral immunity. Cases may be X-linked, autosomal recessive, or autosomal dominant. Because of the severe lack of normal immune function, patients with these disorders are at risk for overwhelming infection by multiple types of pathogens: viruses, bacteria, fungi, and protozoa. Patients usually succumb to infection during the first year of life, although bone marrow transplantation (BMT) may result in reconstitution of the immune system in some cases. Patients are particularly susceptible to normally mild viral respiratory pathogens (parainfluenza, adenovirus, respiratory syncytitial virus).4 Reports of fatal, disseminated adenoviral infection in these children have not mentioned the cornea or conjunctiva among the sites of disease, however. Perhaps because of the rapidly fatal nature of SCID, morbidity from opportunistic infections of the eye has not been among the commonly reported features of these syndromes. Infection with herpes simplex virus (HSV), type 1, including ocular involvement, has been reported in an infant with SCID, but no details were given.32 Cytomegalovirus (CMV) retinitis has been reported in three children with SCID33–35; two patients had been treated by BMT, however.33,35 Severe, disseminated CMV infections occur frequently in patients undergoing this procedure; the relationship between the retinal infection and the immunologic defects of SCID in these patients is therefore uncertain. The third patient was a 5-month-old girl who presented with meningismus and lethargy and was found to have leukocoria.34 SCID was diagnosed during the evaluation of the patient's systemic illness. White vitreous humor masses were found in both eyes, as well as retinal necrosis and retinal detachment. A fine needle biopsy of the mass in one eye revealed cells with cytoplasmic inclusion bodies consistent with CMV infection. Serologic test results were consistent with recent CMV infection in the patient and her mother. Despite treatment with intravenous (IV) ganciclovir, she died 1 month later from pneumonia; CMV and P. carinii were found in her lungs.
Nezelof's syndrome refers to the presence, in infants, of cell-mediated immune defects and an embryonic thymus, but normal circulating immunoglobulins; as such, it is a variant of SCID. There has been no consistency in the literature in the use of this term, and, therefore, it is difficult to compare reports of its manifestations. Among five patients with disorders labeled Nezelof's syndrome, Friedlander and associates36 identified S. aureus blepharitis in one and Pseudomonas aeruginosa-related blepharoconjunctivitis in one. The other patients were without apparent ophthalmic disease. A patient with Nezelof's syndrome and tuberculous meningitis did have optic atrophy among her constellation of findings.37
OTHER DISORDERS OF T AND B LYMPHOCYTES.
Bare lymphocyte syndrome consists of a group of defects that lead to defective expression of Class I or Class II MHC molecules. Candidal chorioretinitis has been reported in a patient with this disorder, but the patient had other, well-established risk factors for disseminated candidiasis, including indwelling catheters and systemic use of broad-spectrum antibiotics.38 The infection cleared with appropriate antifungal therapy; therefore, the effect, if any, of the specific immune defect on the infection cannot be assessed.
Immunodeficiencies Associated With Other Major Defects
WISKOTT-ALDRICH SYNDROME.
Wiskott-Aldrich syndrome is a sex-linked congenital immunodeficiency disease with both humoral and cellular disorders. Patients are also thrombocytopenic at birth. The involved gene, designated WASP (for Wiskott-Aldrich syndrome protein), is expressed in hematopoietic cells and is associated with the cytoskeletal reorganization of activated platelets and lymphocytes. The syndrome is characterized by deficiencies in IgM and elevations in IgA and IgE. Patients develop eczema; thrombocytopenia; and recurrent infections, including laryngitis, bronchitis, gastroenteritis, pyoderma, sinusitis, and otitis media. Infection is caused most often by pyogenic bacteria, but patients with this syndrome are also susceptible to various fungi, viruses, and protozoa. Kaposi varicelliform eruption, a disseminated form of cutaneous HSV infection, may occur. Such infections become apparent by 6 months of age, and susceptibility to infection increases with age. Death often occurs in the first decade of life because of bleeding or infection.
Several ophthalmic disorders are associated with Wiskott-Aldrich syndrome.39,40 Patients may develop ocular surface and adnexal disease, including eczema on the eyelids; blepharoconjunctivitis and pannus formation associated with molluscum contagiosum virus (MCV) lesions; and recurrent, sometimes bilateral, HSV keratitis. Recurrent periocular HSV infections have been reported.41 In many reported cases, the specific pathogens responsible for conjunctivitis are not mentioned. Marginal infiltrates and episcleritis also have been reported; these findings have been attributed to an altered immune response to infectious agents. Papilledema and oculomotor disorders have been attributed to intracranial lesions. Hemorrhage, both intraocularly and on the ocular surface, may occur. In a review of reported cases, Podos and associates39 found that ophthalmic disease (excluding eczema of the eyelids) had been mentioned in 18 of 80 cases. Fifteen patients had infections and five had bleeding of the conjunctiva, periorbital tissues, retina, or into the vitreous humor. Excision of MCV lesions by expression and curettage should be performed with caution in these patients because of the increased risk of hemorrhage.
ATAXIA-TELANGIECTASIA.
Ataxia-telangiectasia is a rare, autosomal recessive disorder characterized by neurologic abnormalities, including cerebellar ataxia, vascular changes of the conjunctiva and skin, recurrent respiratory infections, and malignancies, especially lymphomas. The gene responsible for ataxia-telangiectasia, termed ataxia-telangiectasia muted (ATM), has been mapped to chromosome 11 (11q). Its gene product is a protein kinase involved in cell cycle regulation and deoxyribonucleic acid (DNA) repair.42 Defects in DNA repair, cell cycle regulation, and chromosomal instability may play a role in the observed increase in lymphoid malignancies, as well as in abnormal development of the thymus.
Patients have alterations in both cellular and humoral immune functions; markedly decreased IgA and IgE levels are often present. The disease is progressive, with gradual loss of immunologic and neurologic function. Patients may become mentally retarded. They usually die as a result of this disease before 20 years of age.
Conjunctival vascular changes and eye movement disorders constitute the ophthalmic manifestations of this syndrome.43–45 Dilated venules and irregular vascular segments in the horizontal exposure area give the eye an injected appearance. These findings appear in most people with ataxia-telangiectasia between the ages of 2 and 7 years. These vascular changes have no functional importance. Similar vascular changes occur on the nose, ears, and extremities.
The disorder is associated with various eye movement disorders involving systems that stabilize images on the retina.46 Abnormalities include difficulties in fixation, pursuit, and convergence; nystagmus; an increased delay time for initiation of voluntary horizontal and vertical saccades; hypometria of voluntary saccades (an abnormality in the initiation of the fast component of involuntary saccades induced by optokinetic or vestibular stimuli, with deviation of the eyes in the direction of the slow component, rather than in the direction of the fast component, as in normal study subjects); and decreased slow component velocity of optokinetic nystagmus.44,46 Those affected may have head thrusts when making horizontal refixations, in an attempt to redirect the eyes for fixation on objects of interest; such thrusts are identical to those seen in people with congenital oculomotor apraxia. Visual acuity, pupillary reaction, and fundi are normal.
Other Lymphocyte Disorders
There are many other incompletely characterized disease states that include immunodeficiency among their findings. Those diseases with defects in cell-mediated immunity make the host susceptible to a broad array of infectious diseases caused by bacteria, viruses, fungi, and protozoa. They include opportunistic infections with organisms that generally are commensal to nonimmunocompromised hosts, such as P. carinii and Candida albicans. Devastating, disseminated viral infections by members of the herpesvirus family, adenovirus, rotavirus, enteroviruses, parainfluenza virus, and respiratory syncytial virus can occur. Atypical mycobacterial infections are common and may be widely disseminated.
CHRONIC MUCOCUTANEOUS CANDIDIASIS.
Persons with chronic mucocutaneous candidiasis may have subtle defects both in cell-mediated immunity and in specific antibody responses to Candida sp.47,48 These individuals have severe skin and mucous membrane infections, which can include ulcerative candidal blepharitis and candidal keratoconjunctivitis.49 Photophobia from keratopathy may be the first symptom of disease, occurring at an early age. Findings may include fine punctate erosions of the upper cornea, superficial pannus, and superficial stromal infiltration with scarring. Easty9 believes that these findings are due to bacterial, rather than fungal, infections, but organisms are rarely identified.
These immunologic defects rarely result in candidemia or systemic candidiasis, however. Although people with this disorder have not been reported to develop endogenous candidal chorioretinitis or endophthalmitis in the absence of other predisposing factors, deep tissue infection, including central nervous system (CNS) involvement, has been reported.50–52
Chronic mucocutaneous candidiasis may occur as an isolated disorder, or it may be one of multiple disorders in several different syndromes. Endocrine disorders are common, being seen in perhaps half of patients.53 In 1962, Gass54 reported that keratoconjunctivitis was a frequent initial finding in young children with multiple endocrine deficiency, autoimmune disease, and candidiasis syndrome, a disorder characterized by idiopathic hypoparathyroidism and adrenal insufficiency. Organisms were isolated from the conjunctiva in only one of these cases; it was suggested that the conjunctival disease was an immunologic reaction to fungal antigen. Wagman and associates55 reported a series of 16 patients with the syndrome, 4 of whom had keratitis, anterior stromal vascularization, and scarring. A report of two siblings with this disorder mentioned the additional findings of eyelash and eyebrow loss in both, and small diffuse keratic precipitates in one.56 Wong and Kirkpatrick57 reported clinical improvement in one patient with this syndrome, who was treated with allogeneic lymphocytes and administration of transfer factor and amphotericin B.
Chronic mucocutaneous candidiasis is also part of a syndrome that includes keratitis, ichthyosis, and deafness.58 The precise immunologic defect has not been identified, and evidence of chronic candidiasis has been found in only approximately 20% of cases. Corneal vascularization apparently occurs in most patients with this syndrome, and loss of eyelashes and eyebrows is common. Cowan and associates59 described a family in which three siblings had defects in lymphocyte function and antibody production that they attributed to a defect in biotin metabolism. Clinical disorders included CNS dysfunction and severe mucocutaneous candidiasis, with blepharoconjunctivitis and corneal ulceration. Although candidal infections usually are associated with defects in cellular immunity, antibody defects, such as occurred in these patients, may play a role in their development as well.49
PRIMARY PHAGOCYTIC DYSFUNCTION
Neutrophils, monocytes, and eosinophils comprise the nonspecific phagocyte system, providing immunologic defense against pyogenic bacteria, such as S. aureus, and fungi, such as Aspergillus sp. To accomplish this role, phagocytes are capable of many sequential functions, including response to chemotaxis, adhesion, opsonization, ingestion, and intracellular microbial lysis. Defects in one or more of these functions can result in chronic or recurrent infections that are refractory to antibiotic therapy. Targets of infection include skin, mucous membranes, lung, bone, lymph node, and components of the reticuloendothelial system.
Disorders of phagocytic function can occur in combination with other immune defects. Wheeland and associates60 described a father and son with chronic purulent blepharitis and secondary corneal ulceration who had abnormal neutrophil function and cellular immunodeficiency. Their disease did not fit other well-established syndromes.
Chronic Granulomatous Disease
Chronic granulomatous disease (CGD) is an inherited disorder characterized biochemically by an inability of phagocytes to use molecular oxygen for generation of important microbicidal byproducts, such as hydrogen peroxide, hydroxyl radicals, and superoxide anions, thus making those affected people susceptible to recurrent life-threatening infections. X-linked recessive and autosomal recessive inheritances are seen. The genetic heterogeneity of the disease primarily reflects different mutations in the genes for the four subunits of reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. The diagnosis can be pursued by in vitro testing of phagocyte function, including measurements of the respiratory burst by chemiluminescence or by measuring the ability of an individual's phagocytes to kill catalase-positive bacteria in vitro.
CGD is characterized by recurrent and often chronic infections involving the lungs, lymph nodes, skin and soft tissues, liver, GI tract, bone, and brain. The pathogens involved most often in the disease are S. aureus, Aspergillus sp., Chromobacterium violaceum, Pseudomonas cepacia, and Nocardia sp. Most of the pathogens are catalase-positive microorganisms. Catalase-positive microbes destroy the hydrogen peroxide they generate. CGD phagocytes are defective in hydrogen peroxide generation and are deprived of an alternative source of hydrogen peroxide by which their antimicrobial function could be maintained. Infections with catalase-negative microbes, such as S. pneumoniae, are rare in people with CGD.
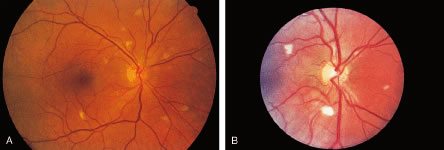
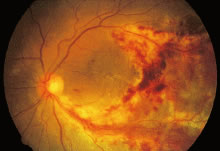
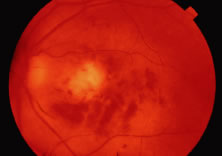
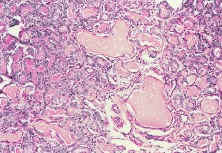
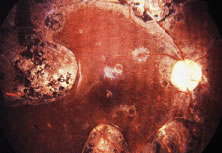
The ocular signs of CGD fall into two groups: recurrent blepharokeratoconjunctivitis and chorioretinal lesions61–64 (Fig. 1). Focal, pigmented chorioretinal lesions appear to be a common finding of CGD. Goldblatt and associates64 examined 38 patients with CGD and 36 of their family members. They found that nine of 38 affected children had chorioretinal lesions, as did three asymptomatic carriers of the gene.64 The lesions are usually perivascular or peripapillary in distribution and can progress to large areas of chorioretinal atrophy.62–64 They are most often described as punched-out chorioretinal scars. Goldblatt and associates64 described small white foveal lesions similar to drusen in some patients, but the typical chorioretinal lesions have not involved the central macula. Visual field defects may occur, corresponding to the location of chorioretinal scars.63 Histopathologic studies of the chorioretinal lesions show foci of granulomatous inflammation of the choroid and sclera.65 The granulomatous inflammation has been seen to extend through Bruch's membrane,65 and areas of Bruch's membrane may be absent.66 Pigment-filled macrophages in the retina and choroid are similar to those found elsewhere in the body.66 Chorioretinal scars with glial proliferation and areas of retinal pigment epithelium (RPE) hypertrophy and hyperplasia surrounded by atrophy of the RPE and the overlying retina are also found.66 Special stains and cultures of the reported ocular pathology and vitrectomy specimens have not revealed any infectious agents.65–67
|
In an isolated case report,65 a 3-year-old child with CGD, who had been documented to have chorioretinal scars previously, was found to have chorioretinal lesions that appeared to be actively inflamed with an associated mild vitreitis. The lesions remained stable for several months, but after 7 months, his left eye developed a severe anterior uveitis, vitreitis, and exudative retinal detachment. A lensectomy and vitrectomy was performed. Cultures and stains of vitreous humor were negative. The presumed endophthalmitis did not respond to IV ceftazidime, nafcillin, and amphotericin B administered for 4 weeks. The eye was ultimately enucleated; cultures and stains of the enucleated eye did not reveal any infectious agents. It is not clear, however, whether there might have been an abnormal response to an infectious agent that had been eliminated by the therapy he received. Most people with CGD do not have any evidence of active inflammation or infection, however.
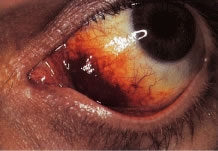
Chronic blepharoconjunctivitis and marginal or punctate keratitis have been reported in patients with CGD, often accompanied by both pannus formation and perilimbal infiltrates.63 These findings are believed to represent an immune reaction to staphylococcal antigens or toxins, rather than a direct invasion of the cornea by organisms. Because it is caused by a catalase-positive bacterium, staphylococcal blepharitis is often a chronic condition in patients with CGD. Regular eyelid hygiene and the selective use of topical antibiotics and corticosteroids has been used to prevent corneal neovascularization and scarring.63
Aggressive and prompt medical and surgical therapy of infections has been important for improving the prognosis of patients with CGD. Granulomata do respond to corticosteroids. The neutrophils of some patients with CGD do respond to interferon (IFN)-γ with a respiratory burst, which may depend on the production of nitric oxide rather than superoxide anions.68 Treatment of CGD with IFN-γ is associated with augmented production of nitric oxide by neutrophils.68 IFN-γ has also been used as a prophylactic agent.69 Although there are reports of cures with BMT, there have been problems with failures to engraft.70
Chédiak-Higashi Syndrome
Chédiak-Higashi syndrome (CHS) is a rare, autosomal recessive disorder, usually occurring in the children of consanguineous parents. It is characterized by partial oculocutaneous albinism, increased susceptibility to both viral and bacterial infections (with both catalase-positive and catalase-negative organisms), anemia, leukopenia, thrombocytopenia, cutaneous ulcers, and peripheral and central neurologic changes, including cerebral atrophy.71 CHS often leads to death in early childhood from infections or, less commonly, from hemorrhage.
Neutrophil abnormalities associated with CHS include neutropenia, reduced bone marrow neutrophil reserves, and reduced neutrophil chemotaxis. Abnormal granules in neutrophils and eosinophils can be seen on routine peripheral blood smears and also are present in other granule-containing cells, such as hepatocytes, renal tubular cells, melanocytes, Schwann's cells, and thyroid cells.72 Ingestion of particulate organisms occurs normally, but the rate of bacterial killing by neutrophils following ingestion is abnormal because of slow release of the abnormally large peroxidase-positive granules, leading to the delayed appearance of myeloperoxidase within phagocytic vacuoles. In addition to the deficient microbicidal activity of lysosomal degradation, natural killer cell activity also has been demonstrated to be abnormal in some cases.73 Heterozygous carriers of CHS have been identified by studies of degranulation in leukocytes.72
Children surviving past early childhood may be afflicted later with a so-called accelerated phase of the disease, characterized by lymphohistiocytic infiltration of the liver, spleen, nerves, and other tissues. Neoplastic transformation of these cells may occur. In addition to antimicrobial chemotherapy for infections, antineoplastic drugs and corticosteroids have been used to treat this accelerated phase.
Oculocutaneous albinism may be a prominent feature of CHS; an ultrastructural defect of melanosomes distinguishes this condition from true forms of albinism. Giant abnormal melanosomes, believed to be result from fusion of smaller abnormal organelles, have been reported. Hair color ranges from blonde to light brown, but most often has been reported as silver or gray.71,74 The iris is translucent with variable color. The amount of pigment in the uveal tract is variable and may be within normal limits. Histologic examination of the eyes also has revealed decreased pigmentation of the RPE, ciliary body, and choroid. These abnormal granules have been observed in monocytes and neutrophils present in the corneal limbus, iris, and choroid.71,75–79 Ultrastructural study of a conjunctival biopsy specimen from a patient with CHS revealed giant intracytoplasmic lysosomal granules in stromal fibroblasts, which are considered to be pathognomonic for CHS; thus, such a biopsy has been suggested as an ancillary diagnostic test in suspected cases of this syndrome.71
Nystagmus and photophobia generally are present, although not in all cases. During the accelerated phase of this disorder, infiltration of the optic disc and central retinal artery has been noted, and papilledema has been reported clinically.71,75–79
COMPLEMENT DEFICIENCIES
The complement system consists of approximately 20 serum proteins that are critical mediators of antigen-antibody reactions. Sequential activation of the complement proteins (complement cascade) is caused by either antibody-antigen interaction, through the so-called classic pathway, or by various stimulants, such as lipopolysaccharide, aggregated immunoglobulin, cobra venom, thrombin, or proteases through the so-called alternate pathway. Specific complement components and complexes of more than one complement protein serve different functions. C3b, for example, promotes immune adherence, phagocytosis, lymphokine production, and antibody-dependent cell-mediated cytotoxicity. C1,4 and C1,4,2,3 complexes aid in neutralization of viruses to which antibodies have attached. The completed complement cascade leads to lysis of viruses, mycoplasma, protozoa, and spirochetes by creating holes in membranes, capsules, or envelopes.
Deficiencies have been reported for each of the proteins that comprise the complement system. Complement deficiencies can result in increased susceptibilities to infections, connective tissue disorders (systemic lupus erythematosus and other autoimmune diseases), or angioedema, depending on which components are involved. For example, C2-deficient patients generally do not have problems with recurrent infections, unless other components, acting later in the cascade, are deficient as well. In contrast, deficiency of C3, which plays an integral role in both the classic and alternate pathways, results in repeated infections. Deficiencies of the later acting components (C5, C6, C7, or C8) have been linked with serious and sometimes fatal infections with N. meningitidis and Neisseria gonorrhoeae. The association between complement deficiency and autoimmune diseases, such as systemic lupus erythematosus, may relate to the host's inability to clear circulating immune complexes.
Although complement has been found in tears,80 cornea,81 aqueous humor,82 and sclera,83 ocular infections have not been reported to be prominent features of complement disorders.9 One should assume, however, that patients will be at risk for ocular infections with the same pathogens seen in nonocular sites. Experimental evidence in animal models shows that the complement system may play a role in protecting the eye against corneal infection84 and endophthalmitis85 caused by P. aeruginosa, an organism whose endotoxins activate the alternate pathway. Complement levels are increased in the vitreous humor of inflamed eyes.86 Patients with complement deficiencies who develop intraocular infections for some other reason may, therefore, have more severe disease than their immunocompetent counterparts.