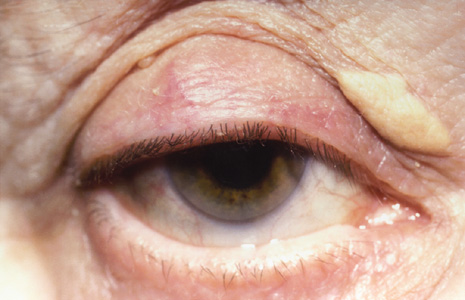
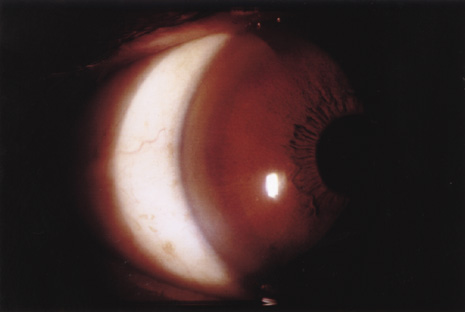
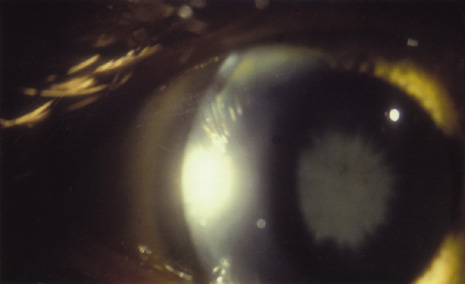
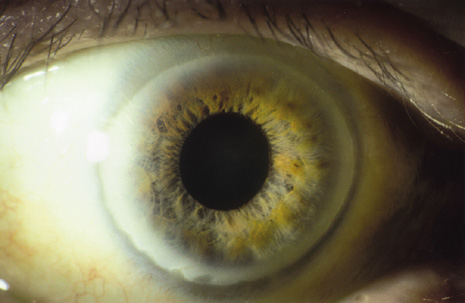
1. Heathcote J: Update on primary biliary cirrhosis. Can J Gastroenterol 14:43–48, 2000. 2. Neuberger J: Liver transplantation for primary biliary cirrhosis: indications and risk
of recurrence. J Hepatol 39:142–148, 2003. 3. Landing BH, Silverman FN, Craig JM, et al: Familial neurovisceral lipidosis. Am J Dis Child 108:503–522, 1964. 4. Kamath BM, Loomes KM, Oakey RJ, et al: Facial features of Alagille syndrome: specific or cholestasis facies? Am J Med Genet 112:163–170, 2002. 5. Bharti AR, Nally JE, Ricaldi JN, et al: Leptospirosis: a zoonotic disease of global importance. Lancet Infect Dis 3:757–771, 2003. 6. Rathinam SR: Ocular leptospirosis. Curr Opin Ophthalmol 13:381–386, 2002. 7. Guidugli F, Castro AA, Atallah AN: Antibiotics for preventing leptospirosis. Cochrane Database Syst Rev 2000:CD001305. 8. Watt G, Padre LP, Tuazon ML, et al: Placebo-controlled trial of intravenous penicillin for severe and late
leptospirosis. Lancet 1:433–435, 1988. 9. Tsianos EV, Hoofnagle JH, Fox PC, et al: Sjögren's syndrome in patients with primary biliary cirrhosis. Hepatology 11:730–734, 1990. 10. Uddenfeldt P, Danielsson A, Forssell A, et al: Features of Sjögren's syndrome in patients with primary biliary
cirrhosis. J Intern Med 230:443–448, 1991. 11. Libert J, Toussaint D: Tortuosities of retinal and conjunctival vessels in lysosomal storage diseases. Birth Defects Orig Artic Ser 18:347–358, 1982. 12. Giraldo P, Perez-Calvo J, Cortes T, et al: Type I Gaucher's disease: clinical, evolutive and therapeutic features
in 8 cases. Sangre 39:3–7, 1994. 13. Smith JA, Chan CC, Goldin E, et al: Noninvasive diagnosis and ophthalmic features of mucolipidosis type IV. Ophthalmology 109:588–594, 2002. 14. Oder W, Grimm G, Kollegger H, et al: Neurological and neuropsychiatric spectrum of Wilson's disease: A
prospective study of 45 cases. J Neurol 238:281–287, 1991. 15. Tauber J, Steinert RF: Pseudo-Kayser-Fleischer ring of the cornea associated with non-Wilsonian
liver disease. A case report and literature review. Cornea 12:74–77, 1993. 16. Schilsky ML: Diagnosis and treatment of Wilson's disease. Pediatr Transplant 6:15–19, 2002. 17. Goyal V, Tripathi M: Sunflower cataract in Wilson's disease. J Neurol Neurosurg Psychiatry 69:133, 2000. 18. Lossner A, Lossner J, Bachmann H, et al: The Kayser-Fleischer ring during long-term treatment in Wilson's disease (hepatolenticular degeneration). A follow-up study. Graefes Arch Clin Exp Ophthalmol 224:152–155, 1986. 19. Esmaeli B, Burnstine MA, Martonyi CL, et al: Regression of Kayser-Fleischer rings during oral zinc therapy: correlation
with systemic manifestations of Wilson's disease. Cornea 15:582–588, 1996. 20. Ferenci P: Diagnosis and current therapy of Wilson's disease. Aliment Pharmacol Ther 19:157–165, 2004. 21. Butler P, McIntyre N, Mistry PK: Molecular diagnosis of Wilson disease. Mol Genet Metab 72:223–230, 2001. 22. Fleming CR, Dickson ER, Wahner HW, et al: Pigmented corneal rings in non-Wilsonian liver disease. Ann Intern Med 86:285–288, 1977. 23. Lipman RM, Deutsch TA: A yellow-green posterior limbal ring in a patient who does not have Wilson's
disease. Arch Ophthalmol 108:1385, 1990. 24. Alagille D: Alagille syndrome today. Clin Invest Med 19:325–330, 1996. 25. Oda T, Elkahloun AG, Pike BL, et al: Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet 16:235–242, 1997. 26. Alagille D, Estrada A, Hadchouel M, et al: Syndromic paucity of interlobular bile ducts (Alagille syndrome or
arteriohepatic dysplasia): review of 80 cases. J Pediatr 110:195–200, 1987. 27. Hingorani M, Nischal KK, Davies A, et al: Ocular abnormalities in Alagille syndrome. Ophthalmology 106:330–337, 1999. 28. Nischal KK, Hingorani M, Bentley CR, et al: Ocular ultrasound in Alagille syndrome: a new sign. Ophthalmology 104:79–85, 1997. 29. Andrews W, Sommerauer J, Roden J, et al: 10 years of pediatric liver transplantation. J Pediatr Surg 31:619–624, 1996. 30. Bruckner R, Batschelet E, Hugenschmidt F: The Basel longitudinal study on aging. Doc Ophthalmol 64:235–310, 1986. 31. Chua BE, Mitchell P, Wang JJ, et al: Corneal arcus and hyperlipidemia: findings from an older population. Am J Ophthalmol 137:363–365, 2004. 32. Chambless LE, Fuchs FD, Linn S, et al: The association of corneal arcus with coronary heart disease and cardiovascular
disease mortality in the Lipid Research Clinics Mortality Follow-up
Study. Am J Public Health 80:1200–1204, 1990. 33. Crispin S: Ocular lipid deposition and hyperlipoproteinaemia. Prog Retin Eye Res 21:169–224, 2002. 34. Eye 3:240–250, 1989. 35. Winder AF: Relationship between corneal arcus and hyperlipidaemia is clarified by
studies in familial hypercholesterolaemia. Br J Ophthalmol 67:789–794, 1983. 36. Civeira F: Guidelines for the diagnosis and management of heterozygous familial hypercholesterolemia. Atherosclerosis 173:55–68, 2004. 37. Barchiesi BJ, Eckel RH, Ellis PP: The cornea and disorders of lipid metabolism. Surv Ophthalmol 36:1–22, 1991. 38. Mahley RW, Rall SC: Type III hyperlipoproteinemia (dysbetalipoproteinemia): the role
of apolipoprotein E in normal and abnormal lipoprotein metabolism. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds). The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill, 1995:1953–1980. 39. Azoulay M, Henry I, Tata F, et al: The structural gene for lecithin: cholesterol acyl transferase (LCAT) maps
to 16q22. Ann Hum Genet 51:129–136, 1987. 40. Idzior WB: Familial LCAT deficiency. Przegl Lek 58:919–923, 2001. 41. Carlson LA, Holmquist L: Evidence for deficiency of high density lipoprotein lecithin: cholesterol
acyltransferase activity (alpha-LCAT) in fish eye disease. Acta Med Scand 218:189–196, 1985. 42. Carlson LA, Philipson B: Fish eye disease. A new familial condition with massive corneal opacities
and dyslipoproteinaemia. Lancet 2:922–924, 1979. 43. Brooks-Wilson A, Marcil M, Clee SM, et al: Mutations in ABC1 in Tangier disease and familial high-density lipoprotein
deficiency. Nat Genet 22:336–345, 1999. 44. Chu FC, Kuwabara T, Cogan DG, et al: Ocular manifestations of familial high-density lipoprotein deficiency (Tangier
disease). Arch Ophthalmol 97:1926–1928, 1979. 45. Guffon N, Souillet G, Maire I, et al: Follow-up of nine patients with Hurler syndrome after bone marrow transplantation. J Pediatr 133:119–125, 1998. 46. Gullingsrud EO, Krivit W, Summers CG: Ocular abnormalities in the mucopolysaccharidoses after bone marrow transplantation. Longer
follow-up. Ophthalmology 105:1099–1105, 1998. 47. Muenzer J: The Mucopolysaccharidoses: A heterogeneous group of disorders with variable
pediatric presentations. J Pediatr 144(Suppl 5):S27–S34, 2004. 48. Kenyon KR: Ocular manifestations and pathology of systemic mucopolysaccharidoses. Birth Defects Orig Artic Ser 12:133–153, 1976. 49. Goldberg MF, Maumenee AE, McKusick VA: Corneal dystrophies associated with abnormalities of mucopolysaccharide
metabolism. Arch Ophthalmol 74:516–520, 1965. 50. von Noorden GK, Zellweger H, Ponseti IV: Ocular findings in Morquio-Ullrich's disease with report of two cases. Arch Ophthalmol 64:585–591, 1960. 51. Kenyon KR, Topping TM, Green WR, et al: Ocular pathology of the Maroteaux-Lamy syndrome (systemic mucopolysaccharidosis
type VI). Am J Ophthalmol 73:718–741, 1972. 52. Goldberg MF: A review of selected inherited corneal dystrophies associated with systemic
diseases. Birth Defects 7:13–25, 1971. 53. Amir N, Zlotogora J, Bach G: Mucolipidosis type IV: clinical spectrum and natural history. Pediatrics 79:953–959, 1987. 54. Traboulsi EI, Maumenee IH: Ophthalmologic findings in mucolipidosis III (Pseudo-Hurler Polydystrophy). Am J Ophthalmol 102:592–597, 1986. 55. Libert J, van Hoof F, Farriaux JP, et al: Ocular findings in I-cell disease (mucolipidosis type II). Am J Ophthalmol 83:617–628, 1977. 56. Quigley HA, Goldberg MF: Conjunctival ultrastructure in mucolipidosis III (pseudo-Hurler polydystrophy). Invest Ophthalmol 10:568–580, 1971. 57. Emery JM, Green WR, Wyllie RG, et al: GM-1-gangliosidosis. Ocular and pathological manifestations. Arch Ophthalmol 85:177–187, 1971. 58. Newman NJ, Starck T, Kenyon KR, et al: Corneal surface irregularities and episodic pain in a patient with mucolipidosis
IV. Arch Ophthalmol 108:251–254, 1990. 59. Kasmann-Kellner B, Weindler J, Pfau B, et al: Ocular changes in mucopolysaccharidosis IV A (Morquio A syndrome) and
long-term results of perforating keratoplasty. Ophthalmologica 213:200–205, 1999. 60. Mullaney P, Awad AH, Millar L: Glaucoma in mucopolysaccharidosis 1-H/S. J Pediatr Ophthalmol Strabismus 33:127–131, 1996. 61. Quigley HA, Maumenee AE, Stark WJ: Acute glaucoma in systemic mucopolysaccharidosis I-S. Am J Ophthalmol 80:70–72, 1975. 62. Cahane M, Treister G, Abraham FA, et al: Glaucoma in siblings with Morquio syndrome. Br J Ophthalmol 74:382–383, 1990. 63. Kakkis ED, Muenzer J, Tiller GE, et al: Enzyme-replacement therapy in mucopolysaccharidosis I. N Engl J Med 344:182–188, 2001. 64. Levy HL, Brown AE, Williams SE, et al: Vitreous hemorrhage as an ophthalmic complication of galactosemia. J Pediatr 129:922–925, 1996. 65. Schweitzer KS: Early diagnosis of inherited metabolic disorders towards improving outcome: the
controversial issue of galactosaemia. Eur J Pediatr 162(Suppl 1):S50–S53, 2003. 66. Beigi B, O'Keefe M, Bowell R, et al: Ophthalmic findings in classical galactosaemia—prospective study. Br J Ophthalmol 77:162–164, 1993. 67. Walter JH, Collins JE, Leonard JV: Recommendations for the management of galactosaemia. UK Galactosaemia Steering
Group. Arch Dis Child 80:93–96, 1999. 68. Badawi N, Cahalane SF, McDonald M, et al: Galactosaemia—a controversial disorder. Screening and outcome. Ireland 1972–1992. Ir Med J 89:16–17, 1996. 69. Poggi-Travert F, Fournier B, Poll-The BT, et al: Clinical approach to inherited peroxisomal disorders. J Inherit Metab Dis 18(Suppl 1):S1–S18, 1995. 70. Folz SJ, Trobe JD: The peroxisome and the eye. Surv Ophthalmol 35:353–368, 1991. 71. Hittner HM, Kretzer FL, Mehta RS: Zellweger syndrome: lenticular opacities indicating carrier status and
lens abnormalities characteristic of homozygotes. Arch Ophthalmol 99:1977–1982, 1981. 72. Cruysberg JR, Wevers RA, van-Engelen BG, et al: Ocular and systemic manifestations of cerebrotendinous xanthomatosis. Am J Ophthalmol 120:597–604, 1995. 73. Moghadasian MH, Salen G, Frohlich JJ, et al: Cerebrotendinous xanthomatosis: a rare disease with diverse manifestations. Arch Neurol 59:527–529, 2002. 74. Dotti MT, Rufa A, Federico A: Cerebrotendinous xanthomatosis: heterogeneity of clinical phenotype with
evidence of previously undescribed ophthalmological findings. J Inherit Metab Dis 24:696–706, 2001. 75. Berginer VM, Salen G, Shefer S: Long-term treatment of cerebrotendinous xanthomatosis with chenodeoxycholic
acid. N Engl J Med 311:1649–1652, 1984. 76. Caruso RC, Kaiser-Kupfer MI, Muenzer J, et al: Electroretinographic findings in the mucopolysaccharidoses. Ophthalmology 93:1612–1616, 1986. 77. Stanesu-Segal B, Evrard P: Zellweger syndrome, retinal involvement. Metab Pediatr Syst Ophthalmol 12:96–99, 1989. 78. Cohen SM, Brown FR 3rd, Martyn L, et al: Ocular histopathologic and biochemical studies of the cerebrohepatorenal
syndrome (Zellweger syndrome) and its relationship to neonatal
adrenoleukodystrophy. Am J Ophthalmol 96:488–501, 1983. 79. Mowat AP: Liver Disorders in Childhood 2nd ed. London: Butterworths, 1987:201–202. 80. Martinez M: Abnormal profiles of polyunsaturated fatty acids in the brain, liver, kidney
and retina of patients with peroxisomal disorders. Brain Res 583:171–182, 1992. 81. Shimozawa N, Suzuki Y, Orii T, et al: Biochemical and morphologic aspects of peroxisomes in the human rectal
mucosa: diagnosis of Zellweger syndrome simplified by rectal biopsy. Pediatr Res 24:723–727, 1988. 82. Moser HW, Moser AE, Singh I, et al: Adrenoleukodystrophy: survey of 303 cases: biochemistry, diagnosis and
therapy. Ann Neurol 16:628–641, 1984. 83. Glasgow BJ, Brown HH, Hannah JB, et al: Ocular pathologic findings in neonatal adrenoleukodystrophy. Ophthalmology 94:1054–1060, 1987. 84. Lyons CJ, Castano G, McCormick AQ, et al: Leopard spot retinal pigmentation in infancy indicating a peroxisomal disorder. Br J Ophthalmol 88:191–192, 2004. 85. Aouburg P, Scotto J, Rocchiccioli F, et al: Neonatal adrenoleukodystrophy. J Neurol Neurosurg Psychiatry 49:77–86, 1986. 86. Rizzo WB, Leshner RT, Odone A, et al: Dietary erucic acid therapy for X-linked adrenoleukodystrophy. Neurology 39:1415–1422, 1989. 87. Miike T, Taku K, Tamura T, et al: Clinical improvement of adrenoleukodystrophy following intravenous gammaglobulin
therapy. Brain Dev 11:134–137, 1989. 88. Aubourg P, Blanche S, Jambaque I, et al: Reversal of early neurologic and neuroradiologic manifestations of X-linked
adrenoleukodystrophy by bone marrow transplantation. N Engl J Med 322:1860–1866, 1990. 89. Aubourg P, Adamsbaum C, Lavallard-Rousseau MC, et al: A two-year trial of oleic and erucic acids (Lorenzo's oil) as
treatment for adrenomyeloneuropathy. N Engl J Med 329:745–752, 1993. 90. Shoulders CC, Brett DJ, Bayliss JD, et al: Abetalipoproteinemia is caused by defects of the gene encoding the 97 kDa
subunit of a microsomal triglyceride transfer protein. Hum Mol Genet 2:2109–2116, 1993. 91. Bishara S, Merin S, Cooper M, et al: Combined vitamin A and E therapy prevents retinal electrophysiological
deterioration in abetalipoproteinaemia. Br J Ophthalmol 66:767–770, 1982. 92. Duker JS, Belmont J, Bosley TM: Angioid streaks associated with abetalipoproteinemia. Arch Ophthalmol 105:1173–1174, 1987. 93. Crocker AC, Farber S: Niemann-Pick disease: A review of eighteen patients. Medicine—(Baltimore) 37:1–95, 1958. 94. Walton DS, Robb RM, Crocker AC: Ocular manifestations of group A Niemann-Pick disease. Am J Ophthalmol 85:174–180, 1978. 95. Bayever E, August CS, Kamani N, et al: Allogeneic bone marrow transplantation for Niemann Pick disease (type
IA). Bone Marrow Transplant 10(Suppl 1):S85–S86, 1992. 96. Bembi B, Comelli M, Scaggiante B, et al: Treatment of sphingomyelinase deficiency by repeated implantations of amniotic
epithelial cells. Am J Med Genet 44:527–533, 1992. 97. Sidransky E, Tsuji S, Martin BM, et al: DNA mutation analysis of Gaucher patients. Am J Med Genet 42:331–336, 1992. 98. Jaime S, Dalmas MF: A case of Gaucher's disease associated with peripheral retinal ischemia. J Fr Ophtalmol 12:461–463, 1989. 99. Tsai P, Lipton JM, Sahdev I, et al: Allogenic bone marrow transplantation in severe Gaucher disease. Pediatr Res 31:503–507, 1992. 100. Fallet S, Grace ME, Sibille A, et al: Enzyme augmentation in moderate to life-threatening Gaucher disease. Pediatr Res 31:496–502, 1992. 101. Barton NW, Brady RO, Dambrosia JM, et al: Replacement therapy for inherited enzyme deficiency: macrophage-targeted
glucocerebrosidase for Gaucher's disease. N Engl J Med 324:1464–1470, 1991. 102. Fine RN, Wilson WA, Donnell GA: Retinal changes in glycogen storage disease type I. Am J Dis Child 115:328–331, 1968. 103. Traboulsi EI, Krush AJ, Gardner EJ, et al: Prevalence and importance of pigmented ocular fundus lesions in Gardner's
syndrome. N Engl J Med 316:661–667, 1987. 104. Aiello LP, Traboulsi EI: Pigmented fundal lesions in a preterm infant with familial adenomatous
polyposis. Arch Ophthalmol 111:302–303, 1993. 105. Burn J, Chapman PD, Delhanty J, et al: The UK Northern Region genetic register for familial adenomatous polyposis
coli: use of age of onset, congenital hypertrophy of the retinal pigment
epithelium and DNA markers in risk calculation. J Med Genet 28:289–296, 1991. 106. Olschwang S, Tiret A, Laurent PP, et al: Restriction of ocular fundus lesions to a specific subgroup of APC mutations
in adenomatous polyposis coli patients. Cell 75:959–968, 1993. 107. Rhodes M, Bradburn DM: Overview of screening and management of familial adenomatous polyposis. Gut 33:125–131, 1992. 108. Shields JA, Shields CL, Eagle RC Jr, et al: Adenocarcinoma arising from congenital hypertrophy of retinal pigment epithelium. Arch Ophthalmol 119:597–602, 2001. 109. Wells AD, McDonnell PJ, Burnand KG: Purtscher's retinopathy in acute pancreatitis. Br J Surg 77:820, 1990. 110. Cohen SY, Gaudric A, Chaine G, et al: Rétinopathie des pancréatites. J Fr Ophtalmol 12:261–265, 1989. 111. Behrens-Baumann W, Scheurer G: Morbus Purtscher. Variationsbreite der klinischen Manifestationen bei 11 Patienten
und Uberlegungen zur Pathogenese. Klin Monatsbl Augenheilkd 198:99–107, 1991. 112. Sanders RJ, Brown GC, Brown A, et al: Purtscher's retinopathy preceding acute pancreatitis. Ann Ophthalmol 24:19–21, 1992. 113. Hollo G, Popik E: Is retinopathy in pancreatitis caused by leukocyte emboli? Acta Ophthalmol (Copenh) 70:820–823, 1992. 114. Grey RH: Visual field changes following hepatic transplantation in a patient with
primary biliary cirrhosis. Br J Ophthalmol 75:377–380, 1991. 115. Welsh BM, Smith AL, Elder JE, et al: Night blindness precipitated by isotretinoin in the setting of hypovitaminosis
A. Australas J Dermatol 40:208–210, 1999. 116. Hopkins DJ, Horan E, Burton IL, et al: Ocular disorders in a series of 332 patients with Crohn's disease. Br J Ophthalmol 58:732–737, 1974. 117. Danzi JT: Extraintestinal manifestations of idiopathic inflammatory bowel disease. Arch Intern Med 148:297–302, 1988. 118. Kochhar R, Mehta SK, Nagi B, et al: Extraintestinal manifestations of idiopathic ulcerative colitis. Indian J Gastroenterol 10:88–89, 1991. 119. Greenstein AJ, Janowitz HD, Sachar DB: The extra-intestinal complications of Crohn's disease and ulcerative
colitis: a study of 700 patients. Medicine—(Baltimore) 55:401–412, 1976. 120. Kaneko E, Nawano M, Honda N, et al: Ulcerative colitis complicated by idiopathic central serous chorioretinopathy
with bullous retinal detachment. Dig Dis Sci 30:896–900, 1985. 121. Billson FA, De Dombal FT, Watkinson G, et al: Ocular complications of ulcerative colitis. Gut 8:102–106, 1967. 122. Pomonis E, Triantafillidis JK, Tjenaki M, et al: Report of Eales' disease and ulcerative colitis in the same patient. Am J Gastroenterol 87:1531–1532, 1992. 123. Lyons JL, Rosenbaum JT: Uveitis associated with inflammatory bowel disease compared with uveitis
associated with spondyloarthropathy. Arch Ophthalmol 115:61–64, 1997. 124. Soukiasian SH, Foster CS, Raizman MB: Treatment strategies for scleritis and uveitis associated with inflammatory
bowel disease. Am J Ophthalmol 118:601–611, 1994. 125. Orchard TR, Chua CN, Ahmad T, et al: Uveitis and erythema nodosum in inflammatory bowel disease: clinical features
and the role of HLA genes. Gastroenterology 123:714–718, 2002. 126. Rosenbaum JT, Smith JR: Anti-TNF therapy for eye involvement in spondyloarthropathy. Clin Exp Rheumatol 20:S143–S145, 2002. 127. Hugot JP, Chamaillard M, Zouali H, et al: Association of NOD2 leucine-rich repeat variants with susceptibility to
Crohn's disease. Nature 411:599–603, 2001. 128. Ogura Y, Bonen DK, Inohara N, et al: A frameshift mutation in NOD2 associated with susceptibility to Crohn's
disease. Nature 411:603–606, 2001. 129. McKay DM: Intestinal inflammation and the gut microflora. Can J Gastroenterol 13:509–516, 1999. 130. Wahl C, Liptay S, Adler G, et al: Sulfasalazine: a potent and specific inhibitor of nuclear factor kappa
B. J Clin Invest 101:1163–1174, 1998. 131. Hampe J, Cuthbert A, Croucher PJ, et al: Association between insertion mutation in NOD2 gene and Crohn's disease
in German and British populations. Lancet 357:1925–1928, 2001. 132. Relman DA, Schmidt TM, MacDermott RP, et al: Identification of the uncultured bacillus of Whipple's disease. N Engl J Med 327:293–301, 1992. 133. Rickman LS, Freeman WR, Green WR, et al: Brief report: uveitis caused by Tropheryma whippelii (Whipple's
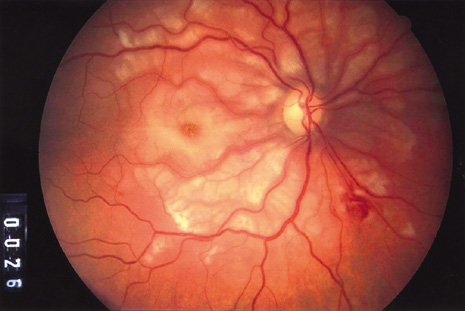
bacillus). N Engl J Med 332:363–366, 1995. 134. Collins ML, Traboulsi EI, Maumenee IH: Optic nerve head swelling and optic atrophy in the systemic mucopolysaccharidoses. Ophthalmology 97:1445–1449, 1990. 135. Harper CG, Giles M, Finlay-Jones R: Clinical signs in the Wernicke-Korsakoff complex: a retrospective analysis
of 131 cases diagnosed at necropsy. J Neurol Neurosurg Psychiatry 49:341–345, 1986. 136. Eggspuhler AW, Bauerfeind P, Dorn T, et al: Wernicke encephalopathy—a severe neurological complication in a clinically
inactive Crohn's disease. Eur Neurol 50:184–185, 2003. 137. Togay IC, Yigit A, Mutluer N: Wernicke's encephalopathy due to hyperemesis gravidarum: an under-recognised
condition. Aust N Z J Obstet Gynaecol 41:453–456, 2001. 138. De La Paz MA, Chung SM, McCrary JA 3rd: Bilateral internuclear ophthalmoplegia in a patient with Wernicke's
encephalopathy. J Clin Neuroophthalmol 12:116–120, 1992. 139. Lengyel D, Weissert M, Schmid L, et al: Eye movement abnormalities as a sign for the diagnosis in Niemann-Pick
disease type C. Klin Monatsbl Augenheilkd 214:50–52, 1999. 140. Grover WD, Naiman JL: Progressive paresis of vertical gaze in lipid storage disease. Neurology 21:896–899, 1971. 141. Horikawa H, Juo K, Mano Y, et al: A case of neurovisceral storage disease with sea-blue histiocyte and severe
horizontal supranuclear ophthalmoplegia. Rinsho Shinkeigaku 30:62–67, 1990. 142. Miller NR: Topical diagnosis of neuropathic ocular motility disorders. In: Miller NR (ed). Walsh and Hoyt's Clinical Neuro-Ophthalmology. 4th ed, vol 2. Baltimore: Williams & Wilkins, 1985:652–784. 143. Schwartz MA, Selhorst JB, Ochs AL, et al: Oculomasticatory myorhythmia: A unique movement disorder occurring in Whipple's
disease. Ann Neurol 20:677–683, 1986. 144. Bradbury JA, Martin L, Strachan IM: Acquired Brown's syndrome associated with Hurler-Scheie's syndrome. Br J Ophthalmol 73:305–308, 1989. 145. Hoft RH, Pflugfelder SC, Forster RK, et al: Clinical evidence for hepatitis B transmission resulting from corneal transplantation. Cornea 16:132–137, 1997. 146. Khalil A, Ayoub M, el-Din-Abdel-Wahab KS, et al: Assessment of the infectivity of corneal buttons taken from hepatitis B
surface antigen seropositive donors. Br J Ophthalmol 79:6–9, 1995. 147. Lee HM, Naor J, Alhindi R, et al: Detection of hepatitis C virus in the corneas of seropositive donors. Cornea 20:37–40, 2001. 148. Holland EJ, Bennett SR, Brannian R, et al: The risk of cytomegalovirus transmission by penetrating keratoplasty. Am J Ophthalmol 105:357–360, 1988. |