FACIAL DEFORMITY SYNDROMES
Mandibulofacial Dysostosis
Mandibulofacial dysostosis (Treacher Collins syndrome) is a syndrome of malformation of structures formed from the first branchial arch, including the zygoma, the ear, the mandible, and the eye (11–13 ). The anomalies may be unilateral or bilateral, and the syndrome may be complete or incomplete. Inheritance is dominant, but expressivity is variable: some patients show the entire syndrome and others only one or two characteristics. Not all patients have a positive family history; there may be more sporadic cases than inherited ones. Chromosomal studies have found no abnormalities.
Nonocular manifestations include hypoplasia of the malar and zygomatic areas. The zygomatic process is occasionally absent and its area depressed; blind fistulas and tongue-shaped extensions of the hairline are also seen in the involved region. The width of the face is reduced. The external ears may be displaced, small, or virtually absent. The external ear canal may be obliterated. The components of the middle ear may be hypoplastic or absent. Mandibular underdevelopment with an associated receding chin is characteristic. Macrostomia is frequent. The palate is high, arched, and often cleft. Malocclusion and malposition of the teeth are the rule. Associated malformations are Klippel-Feil syndrome, spina bifida, aortic coarctation, anomalies of the extremities, and mental retardation.
The most common ocular findings are colobomas in the outer third of the lower eyelids. Other characteristic features include an antimongoloid slant of the palpebral fissures (with the outer canthi lower than the inner canthi) and irregularity or partial or total absence of the lower lid lashes. Microphthalmia, iris colobomas, and absence of lower lacrimal puncta or meibomian glands have occasionally been findings.
Oculovertebral Dysplasia
Oculovertebral dysplasia (Weyers-Thier syndrome) is a unilateral variant of mandibulofacial dysostosis; it is rarer than the more typical forms of the Treacher Collins syndrome (14–16). It is characterized by three classes of deformities: those involving the face, those involving the spine and ribs, and those affecting the eye. The facial malformation, usually unilateral, results mostly from maxillary dysplasia. Thus, there is facial asymmetry, malformation of the superior alveoli, and dental malocclusion. On the affected side, other findings are a low-set and rudimentary external ear, an area of alopecia behind the affected ear, enlargement of the buccal commissure with unilateral macrostomia, and absence of the mandibular ramus. There may be faulty segmentation of the spine and rib anomalies. The hand on the involved side may be deformed.
Ocular findings are unilateral, and they include anophthalmos, cryptophthalmos, and microphthalmos. Weyers and Thier subdivided this syndrome into two types, i.e., a typical unilateral mandibulofacial dysostosis and an oculoauricular form.
Pierre Robin Syndrome
The diagnostic triad in this syndrome consists of micrognathia, glossoptosis, and cleft palate (17). In the newborn with this syndrome, breathing is compromised from pressure of the base of the tongue on the epiglottis. During the second month of embryonal life, closure of the palate normally coincides with rapid mandibular growth. If mandibular growth does not occur (as is the case in the Pierre Robin syndrome), the tongue remains between the palatine processes, and a cleft palate persists. Sixty to eighty percent of infants with micrognathia and glossoptosis have cleft palates. If treatment is not prompt and adequate during earliest infancy, death may occur from asphyxia, vasovagal reflex, inhalation pneumonia, or inanition. Treatment consists of placing the infant in a prone position, feeding through a nasogastric tube, suturing the tip of the tongue to the lower lip, and performing tracheotomy when required.
The prognosis is good if early childhood can be survived. Micrognathia becomes less prominent as the child grows older: by age 5 or 6 these children appear fairly normal. Most cases are sporadic, but, occasionally, familial proclivity has been described.
Associated malformations are aural anomalies, congenital heart disease, and malformation of the extremities. Intelligence is usually normal.
Ocular involvement occurs in approximately half of the patients (18, 19). These include, in order of decreasing frequency, esotropia, congenital glaucoma, retinal detachment, and severe myopia. Also noted have been purulent conjunctivitis in the newborn, posterior subcapsular cataracts, and irregular pupils. Early eye examination should be directed to the detection of glaucoma and retinodialyses with detachments.
Oculoauriculovertebral Dysplasia
Oculoauriculovertebral dysplasia (Goldenhar's syndrome, oculoauricular dysplasia) was first reported by Goldenhar in 1952 (20). It has many of the features of mandibulofacial dysostosis as well as epibulbar dermoids and vertebral anomalies. Together with the Treacher Collins syndrome (mandibulofacial dysostosis) and the Pierre Robin syndrome (micrognathia, glossoptosis, and cleft palate), it is probably a variant of a first branchial arch syndrome. Goldenhar's syndrome exhibits auricular, vertebral, and facial defects (21, 22). There may be microtia (small ears) with or without occlusion of the external ear canal. Preauricular fistulas and appendages are the rule; they occur in 70% of patients (Fig 29-1 ). Block-shaped vertebrae and hemivertebra are especially common in the cervical spine, and they are sometimes combined with occipitalization of the atlas. There may be irregularities in the thoracic, lumbar, and sacral regions of the spine. The skull and face may be underdeveloped as a result of mandibular and malar hypoplasia. High or cleft palate, cleft lip or tongue, micrognathia, and dental anomalies are also seen. No abnormalities have been found in chromosomal studies. There is no familial incidence in 90% of cases. Related systemic defects are usually cardiovascular; rarely, there is a genitourinary defect, hydrocephalus, or mental retardation.
|
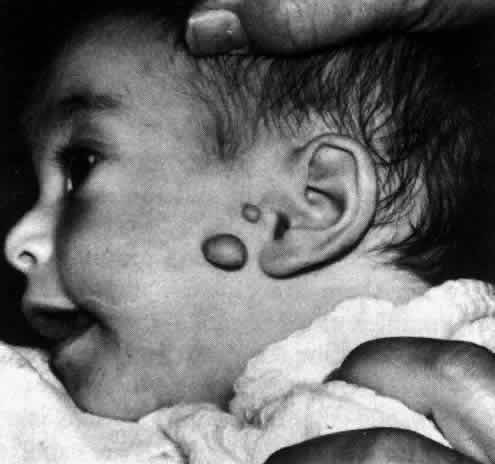
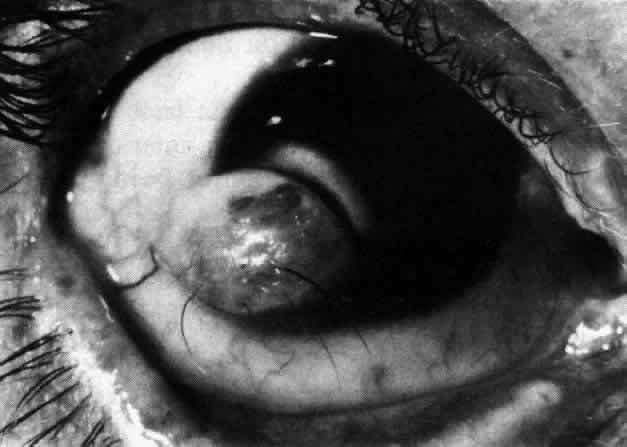
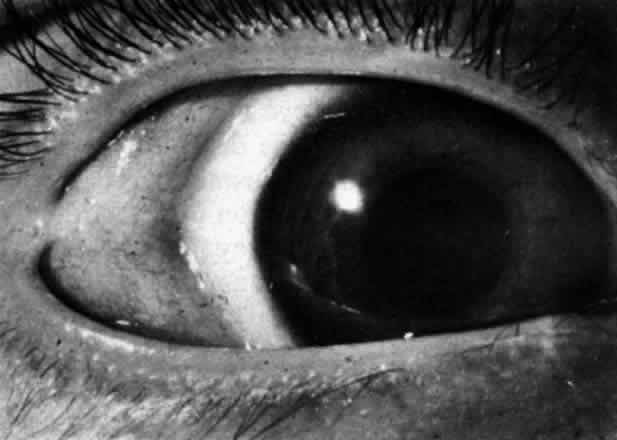
The three most frequent ocular findings are epibulbar dermoids, lipodermoids, and colobomas of the upper lids. The epibulbar dermoids are usually unilateral, are seen at the inferotemporal limbus, and are present in 76% of patients (Fig 29-2). Lipodermoids tend to be superotemporal, are found in 47% of patients, and are sometimes contiguous to an epibulbar dermoid (Fig 29-3 ). Upper lid colobomas are usually unilateral at the junction of the middle and the inner third of the lid, and they are found in 24% of patients. Less frequent ocular findings are Duane's syndrome, dacryocystitis, anophthalmos, microphthalmos, microcornea, iris coloboma, anisocoria, and cataracts.
|
|
Oculomandibulofacial Dyscephaly
Oculomandibulofacial dyscephaly ( Hallermann-Streiff syndrome) is a syndrome of multiple congenital anomalies of the head and face, which gives affected patients a birdlike appearance (23–25). The brow and parietal areas are prominent and the occiput is flat. The sutures and fontanelles remain open at least through infancy and frequently for many years. Size at birth is normal, although dwarfism develops during childhood. In the newborn, the tip of the nose is small and has a beaked appearance; older patients have a long thin nose and receding chin. There is frequently atrophy of the skin of the face. The hair is thin and sparse, with areas of baldness. The mouth and tongue are small, and the palate is high. Dentition is irregular; some teeth never form. Malocclusion is common. There may be anterior displacement of the temporomandibular joint. The ears are low set. The testicles are usually small and undescended. The joints are often hyperextensible, especially the knees. One in 6 patients shows mental retardation. Of the approximately 60 cases reported, familial occurrence has been infrequent. There are no definite chromosomal abnormalities.
Ocular findings include microphthalmos and microcornea, congenital cataracts, antimongoloid slant of the lids, blue scleras, pupillary membranes, nystagmus, strabismus, and retinal anomalies. Eyelashes and eyebrows often are sparse or absent. Orbits are small with reduced interorbital distance.
Facial Hemiatrophy
Facial hemiatrophy (Perry-Romberg syndrome) is a progressive and destructive disease with onset in preadolescence in 75% of patients. It most often affects the face, but there may be extension to the neck, shoulder, trunk, and extremities (26, 27). The brain may also show hemiatrophy, and homolateral migraine and contralateral epilepsy are frequently encountered. Hemiatrophy of fat and subcutaneous tissue is usually the presenting sign in the disease. When the disorder is well developed the appearance is striking: the affected half of the face is sunken and wrinkled. The atrophy frequently involves the soft palate and tongue on the affected side. The maxillary, malar, and palatine bones are smaller and flatter than on the normal side. The external ear may be shrunken and may show appendages. Alopecia and whitening of the hair of the head are manifest early. There may be a localized scleroderma of the forehead near the midline in the form of coup de sabre. Also seen on the affected side are vitiligo, moles, nevus flammeus, and white eyelashes.
Ocular involvement occurred in one series in 19 of 148 patients. Horner's syndrome is fairly common. Enophthalmos, mydriasis, heterochromia iridis, blepharophimosis, ptosis, eye muscle palsies, and nystagmus have been reported on the involved side. A congenitally cystic eyeball has been noted.
After a progressive and irritative stage, the disease may remain unchanged for many years. Some of the cases are familial, and a recessive inheritance has been suggested. Some believe that facial hemiatrophy is related to a disturbance of the sympathetic nervous system.
CRANIAL AND CRANIOFACIAL DEFORMITY SYNDROMES
The Craniostenoses
The craniostenoses include a heterogeneous group of cranial anomalies characterized principally by the premature closure of one or more suture lines in the developing calvarium (28–30). The resulting defect is a function of (1) which suture or sutures are closed, (2) the time in development at which closure takes place, and (3) the extent of closure. The clinical signs and symptoms produced by the craniostenoses depend on the degree of cerebral and foraminal compression that occurs as a result of the bony deformities.
The principal groupings include the following:
- Oxycephaly (tower skull, turricephaly). This condition is due to premature
closure of both the transverse and coronal sutures; the result is
a high, wide head with a short anteroposterior diameter. Because the head
is very high, the highest point being near the bregma, the name “tower
skull” has been applied to this anomaly. It usually appears
sporadically, although a familial incidence has been reported.
- Scaphocephaly. This condition occurs secondary to premature closure of
the sagittal suture, which results in a head with a very long anteroposterior
diameter, a small transverse diameter, and a short height. Scaphocephaly
may be inherited or sporadic, and when inherited it may be
either autosomal dominant or recessive.
- Plagiocephaly. This results from the unilateral closure of the coronal
suture. The head appears asymmetrically enlarged on the side of the unfused
suture.
- Acrocephaly. The acrocephalic head is formed by premature closure of the
anterior sagittal suture; this causes the posterior portion of the skull
to bulge upward, leaving a sloping head with a very posterior summit.
- Miscellaneous. Any cranial suture may close prematurely, resulting in the
anomaly predicted by the anatomy of the skull.
The signs and symptoms produced by the craniostenoses are functions of the abnormal anatomy of the skull. Although prematurely closed sutures are closed at birth, their effect is not fully appreciated until the cranium starts to grow anomalously. The sutures are usually palpable and ridged. Diagnosis is readily apparent from the appearance of the patient, and radiologic studies are confirmatory. Occasionally, defects of the nasopharynx and palate are present concomitantly. Secondary cerebral atrophy and an increased incidence of retardation are probably results of cerebral compression. The symptoms of this group of disorders are mostly referable to the central nervous system. Increased intracranial pressure (occasionally intensified at puberty) may cause headaches, nausea, vomiting, and dizziness. Seizures are particularly common with oxycephaly. Other reported neurologic anomalies include pyramidal tract dysfunction, anosmia, abducens palsy, trigeminal neuralgia, decreased hearing, and poor balance.
The eyes may be involved either as a direct consequence of the disease or by an associated anomaly. Optic atrophy may arise from any one of three causes. Primary optic atrophy may develop due to stretching of the optic nerves, which occurs when the brain is displaced by the cranial anomaly, or when the optic foramens are reduced in size by anomalous bone formation. Secondary optic atrophy may follow chronic papilledema. Exophthalmos may develop when the orbits are abnormally formed and shallow. Strabismus, especially exotropia, is common. Nystagmus, colobomas, and myelinated nerve fibers have also been reported.
The treatment of all these disorders is primarily surgical and is aimed at cosmetic repair of the cranial vault and decompression of the brain. Compression of the optic nerves may necessitate surgery in the area of the optic foramens.
Crouzon's Syndrome
Crouzon's syndrome (craniofacial dysostosis) was first described in 1912 by Crouzon. It is a condition of widespread anomalies of the skull and facial bones (31–33). Inheritance is autosomal dominant, and there are a high degree of penetrance and variable expressivity. However, many of the reported cases occur sporadically. There is an equal incidence in males and females. The condition is easily detectable by the patient's facial appearance and the characteristic x-ray findings.
The head is brachycephalic, and there is a wide transverse diameter. (Although this is the most common anomaly in Crouzon's syndrome, the specific craniofacial anomaly depends on which suture lines are prematurely fused. Brachycephaly is seen often as a result of coronal and lambdoidal synostosis.) The maxilla and zygomatic arch are underdeveloped. The chin and mandible are normal, as is the lower part of the nose. The root of the nose, however, is broad, flat, and depressed. As a result of the midfacial hypoplasia, the normal parts of the face, i.e., the tip of the nose and the chin, appear prominent. The ears are likely to have large lobes, while maldeveloped auditory canals and middle ears frequently lead to impaired hearing (Fig 29-4).
|
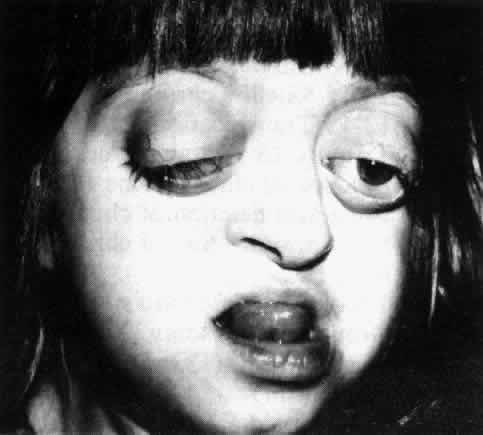
The teeth are irregular and often malformed, and the palate is frequently high and arched.
Central nervous system anomalies consist principally of mental retardation, which is fairly common, and increased intracranial pressure due to extensive synostosis.
When the characteristic findings of Crouzon's disease are combined with acrosyndactyly, the resulting condition is known as “Kephalodactyly,” as described by Vogt in 1933.
The ocular findings are principally a result of increased intracranial pressure and bony malformations of the skull and orbits. Optic nerve compression and dysfunction arise as a result of either chronic papilledema, obliteration of the optic foramens by bony anomalies in the sphenoid bone, or stretching of the optic nerves when the brain is displaced by the abnormal calvarium. Exophthalmos due to abnormally shallow orbits, hypertelorism, exotropia, and antimongoloid palpebral fissures are other ocular anomalies.
The treatment of Crouzon's disease is surgical and is aimed at restoring a satisfactory appearance and normalizing intracranial pressure.
Platybasia
In 1876, Virchow gave the first clear anatomic description of platybasia (basilar impression) of the skull. It is defined as an invagination of the cervical spine into the posterior fossa of the skull. This may occur with great degrees of variation, ranging from mild compression of the atlas and axis toward the foramen magnum to a pronounced form in which the upper vertebral bodies may be fused and completely invaginated into the posterior fossa (34–36). The pons, medulla, and cerebellum may be mechanically distorted and the cranial nerves put on stretch. The upper spinal cord may likewise be deformed, and abnormalities in the circulation of the cerebrospinal fluid are commonly present.
The signs and symptoms of platybasia are highly variable in degree and depend on the extent of the anatomic deformities. Symptoms usually appear in the second to fourth decades, often precipitated by relatively minor cervical or cranial trauma. Presenting manifestations may be headache, cervical pain, cranial nerve disturbances, gait irregularities, limb paresthesias or weakness, or, rarely, blurred vision.
Four distinct insidiously progressive neurologic syndromes may develop either singly or in combination. The first is hydrocephalus, with increased intracranial pressure secondary to abnormalities in cerebrospinal fluid dynamics. Visual loss may ensue as a result of chronic intracranial hypertension. Secondly, a syndrome of cerebellar degeneration may develop as a result of mechanical distortion of posterior fossa structures. Progressive ataxia, nystagmus, and unsteadiness of gait result. The third syndrome is one of chronic spinal cord compression; clinically, it resembles syringomyelia. Manifestations are loss of pain and temperature senses, spasticity, weakness, and atrophy of the limbs. Finally, the syndrome referable to the lower cranial nerves may develop when these are chronically put on stretch and become ischemic. The cranial nerves involved are principally those originating in the medulla.
Diagnosis is made by first noting the patient's physical deformity, viz, a short neck and an occiput which practically rests upon the upper vertebral bodies. Passive motion of the neck is frequently difficult and painful. Pyramidal tract abnormalities, ataxia, and sensory abnormalities (especially those referable to the posterior columns ) may all be evident. Palsies of cranial nerves V, VI, VII, VIII, and IX are present in varying degrees, and loss of arm and hand coordination is frequently seen. A peculiar respiratory disorder develops, in which there is intermittent slowing of respiration to seven or eight breaths per minute. The diagnosis, however, must family rest on radiographic criteria. The chief radiologic criterion is the degree of displacement of the upper vertebral bodies above Chamberlain's line, which is a straight line drawn from the hard palate to the posterior base of the skull..
Ocular findings are primarily a result of the neurologic deficit induced by the anomaly. Nystagmus is the most characteristic feature and may be of any type. Especially localizing is the presence of downbeat nystagmus, which by itself frequently points to a lesion at the level of the foramen magnum. Absence of optokinetic nystagmus and asymmetric optokinetic nystagmus have also been noted, although explanation of the exact anatomic mechanism for this is lacking. Skew deviation may occur, probably as a result of distortion of the cerebellum and its connections, while a form of oculomotor dysmetria has also been described. Finally, chronic papilledema and secondary optic atrophy develop fairly frequently and may be present in either early, intermediate, or late stages of the disease.
The treatmentof platybasia is surgical decompression of the posterior fossa.
It must be emphasized that basilar impression of the skull is seen under many circumstances. Platy-basis may develop ,as an isolated anomaly or along with other malformations of the central nervous system, especially spins bifida, the Klipel-Feil anomaly, the Arnold-Chiari malformation, and meningomyelocele. Central nervous system degenerative conditions such as syringomyelia often coexist with platybasia. Platybasia may be secondary to generalized skeletal disorders, such as Paget's disease or osteogenesis imperfecta, or may be a consequence of metabolic diseases of bone, such as rickets, osteomalacia, or hyperparathyroidism.
Hypertelorism and Hypotelorism
Although hypertelorism was described in several textbooks dating back many centuries, it was Greig, in 1924, who proposed the current scientific explanation. This condition should be defined in rigid terms as a function of the bony structure of the skull and the angle' formed by the medial walls of the two orbits (which are normally parallel). Common facial features of the hypertelorism syndrome include wide separation of the orbits; laterally displaced eyes with an exotropic strabismus; a broad, fiat nasal bridge; and, frequently, a short upper lip, Intracranially, the sphenoid bone is often found to be deformed (28, 37–40).
Idiopathic hypertelorism as described by Greig probably is present in a small and unusual group of patients who have only the pure facial anomaly. This may be inherited as an autosomal dominant or recessive condition, has an unknown sex ratio, and is usually detected early in life.
Most cases of hypertelorism are associated with some generalized syndrome, which is presented under the following categories:
- Chromosomal disorders
- Craniofacial anomalies
- Cleftng syndromes
- Systemic diseases affecting the face and head
Pseudohypertelorism may be due to anomalies of the canthi, nasal root, face, or orbits which give the impression of increased separation of the orbits.
Hypotelorism is a rare facial anomaly usually occurring in conditions not compatible with survival.
The more common causes of these abnormalities are presented below.
- Hypertelorism
- Chromosomal disorders
- Enlarged arms of the 4–5 chromosome
- Short-arm deletion of chromosome 4
- Short-arm deletion of chromosome 5 (cri-du-chat syndrome)
- 6–12 Translocation
- 6–12 Trisomy
- Ring 13–15 (ring D)
- Long-arm deletion of chromosome 13
- 13–15 Satellite chromosome
- 13–15 Translocation
- Trisomy 13 (Patau's syndrome)
- Deletion of chromosome 16
- Long-arm deletion of chromosome 18
- Short-arm deletion of chromosome 18
- Ring 18
- Trisomy 18 (Edward's syndrome)
- Group G monosomy
- Trisomy 21 (Down's syndrome or mongolism)
- The Schmid-Fraccaro syndrome
- XO (Turner's syndrome)
- XXY (Klinefelter's syndrome)
- XXX (Superfemale)
- Enlarged arms of the 4–5 chromosome
- Craniofacial anomalies
- Acrocephalosyndactyly syndromes
- Apert's
- Pfeiffer's
- Others
- Apert's
- Cloverleaf skull ( Kleeblattschädel syndrome)
- Craniofacial dysostosis (Crouzon's disease)
- Craniometaphyseal dysplasia
- Cranio-oculodental syndrome (Chotzens )
- Craniostenoses
- Oxycephaly (turricephaly, tower skull)
- Acrocephaly
- Scaphocephaly
- Plagiocephaly
- Others
- Oxycephaly (turricephaly, tower skull)
- Oculodentodigital dysplasia
- Acrocephalosyndactyly syndromes
- The clefting syndromes
- Cleft lip and palate, acrocephaly, radial aplasia, and absent digits
- Cleft lip and palate, tetraphocomelia, and clitoral or penile enlargement
syndrome (Appelt-Gerken-Lenz, Roberts syndrome)
- Frontonasal dysplasia
- Median cleft face syndrome
- Cleft lip and palate, acrocephaly, radial aplasia, and absent digits
- Systemic abnormalities
- Aminopterin or amethopterin ingestion during pregnancy
- Basal cell nevus syndrome
- Cerebral gigantism
- Cerebrohepatorenal syndrome (Bowen-Zellweger)
- Chondrodystrophia calcificans congenita (Conradi's syndrome)
- Cretinism
- de Lange syndrome
- Dwarfing syndrome of Robinow-Silverman-Smith (fetal face syndrome)
- Familial dysautonomia (Riley-Day syndrome)
- Hallerman-Streiff syndrome
- The happy puppet syndrome
- Hypertelorism, acromegaloid features, and pectus carinatum
- Infantile idiopathic hypercalcemia syndrome
- Leprechaunism
- Lissencephaly syndrome
- Meckel's syndrome (Gruber's)
- Multiple lentigines syndrome
- Noonan's syndrome (male Turner's, Ullrich)
- syndrome
- Seckel-Virchow syndrome (bird-headed dwarfism)
- Thymic agenesis (DiGeorge syndrome)
- Aminopterin or amethopterin ingestion during pregnancy
- Chromosomal disorders
- Apparent Hypertelorism
- Divergence of the eye or eyes secondary to orbital disease
- Polyostotic fibrous dysplasia of bone (Albright's disease)
- Thyroid ophthalmopathy
- Any orbital tumor, pseudotumor
- Polyostotic fibrous dysplasia of bone (Albright's disease)
- Any condition in which epicanthus or pseudoepicanthus is present
- Apparent orbital divergence due to flattened facies (syndrome of cleft
palate, flattened facies, and multiple congenital dislocations)
- Apparent hypertelorism due to a broad, flat nasal root
- Chromosomal defects
- Orofacial-digital syndrome
- Otopalatodigital syndrome
- Smith-Lemli-Opitz syndrome
- Chromosomal defects
- Secondary to nondevelopmental nasal deformity
- Congenital syphilis
- Relapsing polychondritis
- Trauma
- Congenital syphilis
- Apparent separation of the orbits secondary to medial telecanthus
- Acrocephalopolysyndactyly (Carpenter's syndrome)
- The hypertelorism-hypospadias syndrome (patients with this syndrome rarely
have true hypertelorism)
- Schwartz-Jampel syndrome (Aberfeld's)
- Waardenburg syndrome
- Acrocephalopolysyndactyly (Carpenter's syndrome)
- Divergence of the eye or eyes secondary to orbital disease
- Hypotelorism
- Cebocephalia
- Craniotelencephalic dysplasia
- Cyclopia
- Ethmocephalia
- Holoprosencephaly (arrhinencephaly)
- Holotelencephaly
- Trisomy 13 (rare cases)
- Weill-Marchesani syndrome
- Cebocephalia
GENERALIZED SKELETAL DISORDERS
Apert's Disease
First described by Apert in 1906, Apert's disease (acrocephalosyndactyly) is a congenital anomaly affecting bones throughout the body. This, in turn, produces various mechanical defects of the soft tissues in contact with the abnormal bony structures (28, 41–43). Although most cases are sporadic, an autosomal dominant inheritance pattern has been demonstrated in some families. Although demographic studies have not been carried out in all populations, an estimated incidence of 1 in 160,000 live births has been calculated for Anglo-Saxon populations. Males and females are affected equally. Some recent genetic studies have implicated increasing parental (especially paternal) age as a related factor. The condition has been reported in nearly all races, and the intelligence of the affected individuals is usually normal.
The cranial malformation is principally a premature synostosis of the coronal suture and the hypoplasia of the mid base of the skull and the central face. The bones involved in the latter comprise the sphenoethmo-maxillary complex. The sutural synostosis leads to a characteristic oxycephalic skull deformity in which there is an inconspicuous occiput and a high point anterior to the bregma. The forehead is vertically oriented with no slope, and the anteroposterior diameter of the skull is greatly shortened. Neurologic disturbance is a rare consequence of the cranial malformation. These patients are of normal intelligence unless there is neurologic insult. A broad, depressed nasal bridge and relative prognathism are intrinsic parts of the facial anomaly, while posterior cleft palate is occasionally reported.
Other skeletal anomalies most frequently involve the vertebral bodies, the digits, and the limb girdles. A fusion of the fingers is an integral part of the syndrome and characteristically involves the second through fourth digits. The presence of a single fused fingernail is especially characteristic, while involvement of the first and fifth digits is seen occasionally. The syndactyly involving the lower extremities is more likely to include a soft-tissue union of the affected members than that of the upper extremities. The vertebral bodies exhibit varying degrees of fusion, which tends to increase with advancing age. Finally, varying bony abnormalities of the shoulder and pelvic girdles may occur which result in decreased range of motion along with pain and difficulty in active and passive movements.
Many apparently unrelated visceral anomalies have been described; these include a variety of congenital defects of the heart and great vessels, polycystic kidneys and hydronephrosis, esophageal atresia and pyloric stenosis, and thyroid adenomas.
The ocular findings are those secondary to the frequently present cranial anomalies and those which occur in relation to, but not as a direct consequence of, the bony malformations. An antimongoloid palpebral slant with the outer canthi measurably lower than the inner canthi is common. Keratoconus and congenitally absent superior rectus muscles have rarely been reported. Proptosis secondary to an anatomically shallow orbit may lead to severe exposure keratitis. The orbital anomaly is usually caused by an anterior dislocation and an abnormal shape of the sphenoid bone. The most common ocular defect is optic atrophy with resultant visual loss. This may be due to a bony abnormality of the optic foramen in the sphenoid bone or traction on the optic nerve secondary to the cranial maldevelopment. It may also be due to the chronic papilledema which occurs when cerebrospinal fluid dynamics are altered. The orbits are usually divergent in position, and an exotropic strabismus is commonly present.
Surgical intervention is undertaken with three principal aims. First, increased intracranial pressure must be relieved by a cranial decompression procedure. When the optic nerves are compromised within the optic canals, decompressive surgery may also be necessary. Cosmetic surgery of the face and head may be performed, while reconstructive surgery of the hands has a good success rate in the treatment of the syndactyly.
Marfan's Syndrome
Marfan's syndrome (dystrophia mesodermalis congenita—typus Marfanus, arachnodactyly) was alluded to more than 100 years before Marfan, in 1896, put together the skeletal manifestations of the disease which now bears his name. The classic triad of Marfan's syndrome, viz, subluxated lenses, skeletal anomalies, and cardiovascular disease, implies a limited disease in both its ocular and systemic manifestations. Quite to the contrary, Marfan's syndrome involves not only the skeletal and cardiovascular systems but also the lungs, muscles, genitourinary system, and skin as well as affecting nearly every structure of the eye (44, 45, 46 [pp 61–244],47, 48). This autosomal dominant condition has great variability of expression; it affects both sexes equally, is found in all races, and is common enough to be seen several times a year in a busy clinic setting. Although the exact biochemical defect has not been identified, it is evident that the cause must be a basic anomaly of connective tissue or ground substance.
The most striking physical features of a patient with Marfan's syndrome are the musculoskeletal defects. As a rule, patients are tall and have long extremities; their arm span is greater than their total height. Arachnodactyly results in characteristic hands in which the fingers are disproportionately long and thin (Fig 29-5 ). Prognathism and a high arched palate occur occasionally, and the long, thin face is characteristic. Kyphoscoliosis is the most frequently seen vertebral abnormality, while pectus excavatum is one of the most characteristic of all the bony changes. A generalized muscular hypoplasia and hypotony lead to laxity of the joints; occasionally there is contracture. Rocker bottom feet and genu recurvatum are results of joint hyperextensibility with subsequent contracture.
|

Cardiovascular anomalies are found in more than one third of all patients with Marfan's syndrome and are potentially the most serious symptom. Death in patients with Marfan's syndrome is usually due to heart disease. Most pathology is centered about the aortic valve and ascending aorta, in which a degenerative process of the tunica media leads to the formation of dissecting aneurysms. When this type of vascular disease occurs in young patients, Marfan's syndrome must always be suspected. Occasionally, dissection occurs acutely and the patient complains of a tearing pain in the precordium and suddenly develops a new loud murmur. Other valves may also be affected, and the characteristic murmurs of mitral and tricuspid insufficiency are diagnostic. The lungs are the seat of varied anomalies, including abnormal lobulation, cystic malformations, aplasia of parts of the lungs, progressive emphysema, and, occasionally, fibrosis of no apparent inflammatory etiology.
In the genitourinary system, small mobile kidneys may be found and ureteric strictures may be present which predispose to recurrent bouts of pyelonephritis. Epidermal striae are found, and a remarkable lack of subcutaneous fat is a nearly universal occurrence.
Not all patients with Marfan's syndrome are tall and thin with long fingers and pectus excavatum. Patients may appear phenotypically normal with only eye signs, such as dislocated lenses, to suggest the diagnosis.
Myopia, ptosis, megalocornea, and blue scleras have all been reported frequently in these patients. Strabismus is likewise extremely common, but its presence alone does not suggest the diagnosis. The anterior segment structures exhibit both subtle and gross changes, some of which (especially in combination) are extremely suggestive of Marfan's syndrome. The anterior chamber angle exhibits an immature ciliary body with hypoplasia of the muscular elements, a very broad trabecular meshwork, and an inconspicuous Schwalbe's line. Transillumination demonstrates that the iris has a very posterior insertion on the ciliary body, the processes of which extend radially behind the iris much more than usual. The iris tends to show no circumferential ridges, few crypts, and, occasionally, iris root cysts, while iridodonesis occurs when the lens is subluxated. One peculiar abnormality is a hypoplasia of the iris dilator muscle; this causes the pupil to remain small even in response to mydriatic drops. The principal lenticular anomaly is ectopia lentis. This is found in the vast majority of patients with Marfan's syndrome; it is usually bilateral and commonly occurs as a superior or supernasal displacement (Fig 29-6). This is presumably due to poor zonular attachments; for the same reason, lens colobomas may be found. Spherophakia and cataract formation also occur. Various types of retinal degeneration may be seen, including myopic and lattice degeneration and vitreous traction syndromes in the peripheral retina. Both retinal detachment and a peripheral pigmentary retinopathy are common.
|
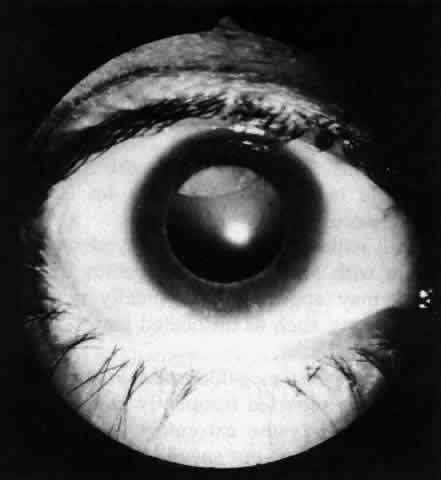
Glaucoma complicating Marfan's syndrome may arise via any one of several mechanisms, including direct pupillary block from anterior lens dislocation, lens luxation into the anterior chamber, and upward or downward lens dislocation resulting in crowding of the anterior chamber angle by mechanical movement of the iris. A chronic form of glaucoma possibly relating to an abnormal insertion of the ciliary musculature into the trabecular meshwork may be seen, as may glaucoma complicating retinal detachment, cataract surgery, or chronic iritis.
The treatment of the nonocular features of Marfan's syndrome is primarily supportive. Occasionally, orthopedic procedures may be undertaken to correct extremity deformities. When there is a dissecting aneurysm of the ascending aorta, the prognosis is poor for cardiovascular surgery.
The main ophthalmic procedure performed in this syndrome is cataract extraction. This is done for cataract formation, lens dislocation, and glaucoma. The procedure is, however, quite complicated in patients with Marfan's syndrome. Virtually all the complications of cataract surgery develop with greater frequency in these patients than in the general population. Vitreous loss and incarceration in the wound, iris prolapse, corneal edema, postoperative hyphema, and persistent postoperative iritis are frequent. Retinal detachment surgery should be performed when needed, using standard techniques. Strabismus repair is indicated under circumstances which would suggest surgery in the general population. Since these patients are of normal intelligence, and since neurologic stigmas are uncommon in Marfan's syndrome, any central nervous system abnormality which does occur should be appropriately investigated.
Approximately 50% of patients with Marfan's syndrome are diagnosed by the ophthalmologist. This is usually because myopia is not adequately corrected by lenses. When a new patient is seen by the ophthalmologist, referral for full medical and orthopedic evaluation is incumbent on the primary physician. The immediate family should also be completely examined.
Weill-Marchesani Syndrome
In 1932, Weill, while describing a series of patients with Marfan's syndrome, noted that 1 of the 8 patients was short in stature and had short swollen fingers as well as dislocated lenses. However, it was not until 1939 that Marchesani reported on the occurrence of the above features in a distinct syndrome which now bears the names of both authors. The Weill-Marchesani syndrome (spherophakia-brachymorphia syndrome, congenital mesodermal dysmorphodystrophy--brachymorphic type) occurs as an autosomal recessive trait and ranks third behind Marfan's syndrome and homocystinuria as a cause for dislocated lenses on an hereditary basis. Males and females are affected equally. Carriers reflect the condition incompletely: they have a short pyknic habitus, but there are no ocular findings. The disease is compatible with normal longevity, and affected patients have normal intelligence. Principal findings are limited to the skeletal system and the eyes (46 [pp 828–892],49–51 ).
The musculoskeletal system shows characteristic brachymorphia, which is marked by short stature, well-developed muscles, a barrel-shaped chest, and strong girdle muscles. The hands are short and spadelike, with stubby fingers, prominent knuckles, and knobby interphalangeal joints. The hands, fingers, and wrists exhibit marked limitation of both active and passive movements. These findings are present from a young age, and, although brachymorphia is characteristic, these patients cannot be classified as true dwarfs. No biochemical abnormality of bone and no specific histologic abnormality of connective tissue have been found. Radiologic changes have been described in the hands, but no specific features have been noted.
The ocular findings are the most prominent and disabling part of the disease. The first ocular abnormality which becomes manifest is usually a moderately high to very high degree of myopia. Even after adequate refraction, these patients often achieve levels of vision in the range of 20/60 or less. The lens is shortened in its horizontal and vertical dimensions and only slightly increased in its anteroposterior dimension and thus approaches sphericity. The small round lens commonly dislocates and may move either down, anteriorly, or posteriorly or may rotate about its axis. Cataract formation is common. It has been shown that after extraction these lenses measure approximately 7 mm in diameter (normal is 9 mm) and are reduced approximately 25% in weight. The most serious consequence of lens dislocation is glaucoma, which may occur via several lenticular mechanisms. Anterior movement causes repeated episodes of pupillary block with secondary angle closure and the development of peripheral anterior synechias. Others have felt that inferior dislocation of the lens may cause increased pressure by either irritation of the ciliary body with overproduction of aqueous or mechanical anterior displacement of the inferior iris with subsequent angle closure in that area. Dislocation of the entire lens into the anterior chamber produces a very severe intractable glaucoma. Other mechanisms proposed for the occurrence of glaucoma include hypoplasia of the ciliary body with mechanical crowding of the angle and one description of an abnormal chamber angle containing numerous pigmented fibers extending from the iris root to the ciliary body trabecular meshwork and Schwalbe's line (49). Examination of the zonules has shown them to be elongated and irregular, and after cataract extraction these fibers may contribute to the development of pupillary block glaucoma. Although pigmentary retinopathy and choroidal degeneration have been described in association with the Weill-Marchesani syndrome, these findings are considered to be unrelated.
The treatment of Weill-Marchesani syndrome is basically directed at various manifestations of the eye disease. Pupillary block glaucoma is best treated with iridectomy. The use of miotics when there is a mobile lens is likely to increase the pupillary block. When lens extraction becomes necessary because of either intractable glaucoma or cataract formation, the procedure must be approached with caution, since complications are common. In one series of 11 intraocular procedures, immediate surgical complications included 2 instances of vitreous loss, 1 case of pupillary block glaucoma, and 1 case of unplanned extracapsular cataract extraction followed by expulsive hemorrhage (50). Visual acuity postoperatively is usually satisfactory except in those patients in whom preoperative glaucoma has resulted in extensive visual loss. Rarely, cases of congenital nystagmus have precluded good postoperative visual results, but surgery must be performed in many cases to control intraocular pressure.
The differential diagnosis of ectopia lentis includes Marfan's syndrome, homocystinuria, hyperlysinemia, sulfite oxidase deficiency, congenital syphilis, trauma, and the Weill-Marchesani syndrome. Some of these are practically nonexistent in clinical practice, i.e., hyperlysinemia and sulfite oxidase deficiency, while the others should be easy to differentiate clinically from the Weill-Marchesani syndrome, due to the characteristic brachymorphic appearance of the latter.
Paget's Disease
Paget's disease (osteitis deformans) was first described in 1877 by Paget and is the most common metabolic bone disease seen in clinical practice. It is usually seen in whites and only rarely reported in orientals. There is no sex predilection, and evidence exists for autosomal dominant, autosomal recessive, and sporadic forms. Age at onset is usually over 50 years, although some patients are in their 30s. The diagnosis should be viewed suspiciously in anyone under the age of 20. One estimate of the prevalence of Paget's disease is that up to 3.3% of the population over 40 years of age show some radiographic evidence of osteitis deformans.
The symptoms of Paget's disease are almost entirely referable to bone; other systems are involved only secondary to compression by abnormal osseous tissue (52, 53 [pp 240–272],54, 55). Although at one time it was felt that an arteriovenous shunt exists in the remaining normal bone and that this results in a high-output cardiac state, the physiologic presence of these shunts has not been verified and is in doubt. The typical clinical presentation is that of an elderly, balding man who has prominent temporal vessels and who complains of pain and tenderness over his shins. The skin may be warm and tender over the slightly bowed tibias. This bowing is usually in an anterolateral direction. When the vertebral bodies as well as the extremities are involved the patient may notice his height gradually decreasing and a severe steady pain in the lower back. Pain and deformity of the lower extremities are almost always greater than that of the upper extremities. Fractures are common, especially in the femur and tibia. Eventually these will heal, but the process is slow and requires immobilization, which in turn leads to a rapid increase in bone resorption.
Involvement and deformity of the skull are common and are the root of many of the neurologic and ophthalmologic complications of Paget's disease. Increasing head size and deafness secondary to bony sclerosis of the auditory system are both present in a high percentage of severely affected patients. Other nerve roots and the spinal cord itself may be compressed, while severe bone deformity within the skull may act as an intracranial mass. Hemorrhage from brittle abnormal bone in the skull may result in either an epidural or a subdural hematoma which may require neurosurgical intervention. Very severe cases involving the upper vertebral bodies and skull may result in a basilar impression syndrome with all its attendant features.
Radiographic signs of Paget's disease vary from osteoporosis circumscripta (the localized thinning and demineralization of bone, especially in the skull) to bizarre and advanced deformities of all the bones in the body. Sclerosis is a common change; it may be demonstrated on x-ray as an increased density of bone which may be bordered by areas of bony thinning. Fine details of vertebral bodies and the bones comprising the base of the skull may be lost, obscured by areas of sclerosis and demineralization. Pathophysiologically, Paget's disease probably represents an abnormally high rate of bone resorption and reformation with a loss of the normal architecture. The two processes are usually accelerated in balance, so there is no net resorption or reformation. However, when there is a fracture and the patient is immobilized, the balance may be shifted, and serum hypercalcemia may occur as a result of net bone resorption.
The only characteristic biochemical abnormality in Paget's disease is a greatly elevated serum alkaline phosphatase level.
The principal osseous complication is malignant degeneration of the abnormal bone. This has been estimated to occur in 5% to 10% of the most severely involved patients and in 1% to 3 % of the patients with less severe involvement. These tumors are usually osteosarcomas, fibrosarcomas, or, quite rarely, giant cell tumors. When they involve the skull they may behave as intracranial mass lesions.
The ocular findings associated with Paget's disease are of two principal types: (1) the intrinsic disorders of the eye associated with osteitis deformans and (2) the effects on the eye produced by the bone disease when it encroaches upon nerves and blood vessels. Primary ocular manifestations of Paget's disease most commonly include choroidal sclerosis and angioid streaks. The precise percentage of patients in whom these manifestations occur has not been determined, but both are seen frequently enough that their presence should not be considered unusual. The choroidal sclerosis is of the senile diffuse type, and the angioid streaks appear much the same as they do in certain associated conditions, e.g., pseudoxanthoma elasticum, sickle cell disease, Ehlers-Danlos syndrome, and familial hyperphosphatemia. Only in Paget's disease and pseudoxanthoma elasticum is it common to find these angioid streaks progressing to disciform degeneration with severe loss of central vision. Cataract formation may be seen in patients with Paget's disease, but this may be nothing more than a coincidence occurring in an aging population.
Manifestations produced by bony interference with nervous impulses result in a much higher ocular morbidity than does any intraocular disease. Spasm of the seventh nerve may develop bilaterally, with associated painful blepharospasm. There may be trigeminal neuralgia which may be similar to any form of tic and which may be associated with lacrimation and a red eye. Extraocular muscle palsies are seen; these may result from compression of nerves in various cranial foramens, or, in the case of the trochlear nerve, bony deformity of the trochlea may cause a mechanical limitation of the superior oblique function. Visual loss due to optic nerve compression may progress slowly and insidiously, causing bizarre visual field defects and eventually resulting in optic atrophy. Rarely, extensive orbital involvement by Paget's disease may cause an increase in orbital venous pressure with a resultant increase in intraocular pressure.
The treatment of Paget's disease has traditionally been supportive, i.e., setting fractures, surgically treating orthopedic problems when necessary, and providing therapy for malignant tumors. Therapy aimed at the basic physiologic abnormality has been proposed. This includes mithramycin and actinomycin D, both antimetabolites; thyrocalcitonin and glucagon; fluorides, magnesium, disodium etidronate, and pyrophosphates (all chemicals aimed at altering bone metabolism); and, finally, radio-therapy. Although many of these have shown promise, none has provided a complete answer.
Osteogenesis Imperfecta
Although originally described late in the 18th century, osteogenesis imperfecta (Van der Hoeve's syndrome, brittle bone disease) was not well described as a clinical syndrome until 1918 by Van der Hoeve. The triad of brittle bones, blue scleras, and deafness (otosclerosis) is basically a disorder of bone matrix and collagen (46 [pp 390–454],53 [pp 162–178],56 ). Osteogenesis imperfecta has an incidence of approximately 1 in 50,000 births, shows no race or sex predilection, and usually has an autosomal dominant inheritance. Autosomal recessive inheritance and sporadic cases also occur but are considerably less frequent. The specific biochemical or enzymatic defect is yet to be identified; however, collagen fibers in the eye have been found to be reduced in periodicity of cross striations and thickness when examined on electron microscopy.
Osteogenesis imperfecta has been divided into two distinct clinical syndromes: osteogenesis imperfecta congenita, which is manifest early in life and results in early death, and osteogenesis imperfecta tarda, which becomes manifest early in childhood and may run a benign, prolonged course. The skeletal defects in the congenita type are usually detectable at birth (or in utero, if antenatal x-rays are taken) and are a direct result of a primary defect in the osteoid and collagen which comprise the bone matrix. The extremities are often deformed, very small, and bent as a result of the bony anomalies. Especially characteristic is the caput membranaceum. The latter is a term used to describe a skull made up of wormian bones which has the radiologic appearance of a flat bone mosaic assembled like a jigsaw puzzle. The calvarium is soft, and death often occurs as a result of intracranial hemorrhage. There may be hydrocephalus due to bony abnormality of the skull. Rib fractures and other rib anomalies are common. The total muscle mass is reduced, and muscular hypotony occurs because of poor tendinous insertions into bones. Radiologically, the long bones show a thin cortex with slender shafts. Should the patient survive, the extremities will remain short and anomalous. Fractures are extremely common and occur with the slightest trauma (e.g., movement in bed, lifting of a child).
The tarda variety is much more irregular in its presentation. When bone disease is present, the first sign is usually a fracture which occurs at 2 to 4 years of age. Fractures are associated with little pain, probably due to the minimal degree of soft-tissue injury. Wormian bones are also present in the skull, and pseudoarthroses are a common finding late in the disease. Scoliosis and other vertebral abnormalities are frequent, and back pain is typical. The skeletal changes in the tarda form are reduced in frequency and severity at puberty and before menopause. Not all patients with osteogenesis imperfecta tarda have gross bony anomalies; in some patients the disease may be limited to relatively minor radiologic changes combined with ocular or aural disease.
Ear findings are an integral part of the syndrome, but deafness is the least common finding in the originally described triad. Symptoms range from tinnitus and vertigo to total deafness, and the finding of soft, chalky bone at the stapedial crura is characteristic. The hearing loss is aggravated by middle ear infections, which occur with greater frequency in these patients than in the general population.
Cutaneous anomalies are seen but are usually subtle. The skin may appear atrophic and overly aged, and scars tend to be wide and lax. Minor trauma may induce fairly large subcutaneous hemorrhage, and capillary fragility is increased. Cardiovascular findings are not a large part of the syndrome, but peripheral artery calcifications may occasionally be seen in the congenita form. Cardiac murmurs, usually of the valvular insufficiency type, are encountered but do not correspond to specific cardiac pathology.
Dental abnormalities comprise a curious and characteristic part of the syndrome. (One form of osteogenesis imperfecta may be limited to the teeth. ) There is yellowish brown or gray discoloration of both the permanent and deciduous teeth, particularly of the incisors. Since the enamel of the teeth is normal and the dentin is abnormal, caries are infrequent, as they are sloughed by the soft teeth. Neurologic findings such as hydrocephalus, cerebellar degeneration, pyramidal dysfunction, and nerve root compression are a result of anomalous bone in contact with nervous tissue. No primary neurologic disease has been demonstrated, and the patients are of normal intelligence.
Ocular findings are varied, but the most prominent and well-recognized finding is the blueish coloration of the sclera due to increased translucency allowing the uveal pigment to shine through. Whether patients with blue scleras and no other findings of osteogenesis imperfecta really have the disease or not is a philosophic question. In pathologic examination, both the cornea and sclera have been found to be reduced in thickness by 50% to 75%, and megalocornea and keratoconus have been coincidentally described. Hypermetropia, zonular cataracts, posterior embryotoxon, and retrobulbar neuritis have also been irregularly reported, while glaucoma occurs even less frequently. A common finding is the appearance of a Saturn ring, which is a comparative whitening of the paralimbal sclera occurring as a result of the lack of pigmented uvea behind the sclera. Optic atrophy may be due to bony compression of the nerve or may develop secondary to chronic papilledema.
Treatment is unsatisfactory in nearly all aspects of the disease. The fractures must be treated in a fairly customary manner, although immobilization should be minimized. Various bone-building diets, consisting of high doses of vitamin C, vitamin D, calcium, phosphorus, and fluorides, have been tried, but the results have been inconclusive. Anabolic steroids seem to decrease the incidence of fractures but do not change the basic course of the disease. Heroic surgical procedures designed to straighten limbs and eliminate pseudoarthroses have met with a modicum of success. The eye disease requires only supportive treatment.
Osteopetrosis
Osteopetrosis (marble bone disease, Albers-Schönberg disease) was first described in 1904 by Albers-Schönberg, a German radiologist. It is a disease of abnormal bone formation which may affect virtually the entire skeleton (46 [pp 809–820],53 [pp 178–193],57, 58). There is no sex predilection, and new patients have been discovered at various ages, from early infancy to adulthood. Two distinct clinical subgroups have been delineated. The first has an autosomal recessive inheritance, becomes clinically manifest very early in life, and is usually lethal. This form has been referred to in the literature as malignant osteopetrosis. The second form has an autosomal dominant inheritance, may not be diagnosed until later in life, and, although causing a high degree of morbidity, only rarely causes early death.
The basic defect in both forms of the disease is one of abnormal bone formation and maturation. Long tubular bones lose their normal cortical and medullary orientation. The vertebral bodies tend to become thickened and sclerosed at their upper and lower portions and demineralized in between. The short round bones of the hands and feet become very brittle, and their character may be hard and fragile or soft and crumbly. Cartilage is found intermingled with the abnormal bone. A characteristic bone-within-bone pattern is commonly found on x-ray. Large flat bones such as the skull and pelvis may be thickened and sclerosed or softened irregularly. When this occurs in the skull, encroachment upon foramens or the brain may result in the development of neurologic symptoms. The sella turcica often becomes distorted, and there may be pituitary dysfunction. Parameters of calcium metabolism have all been found to be normal with the exception of an occasionally increased value for the serum acid phosphatase. Urinary hydroxyproline, an index of bone resorption, has likewise been found to be normal.
The malignant or infantile form is characterized by hepatosplenomegaly and lymphadenopathy; these are presumed to develop secondary to the presence of extramedullary hematopoietic elements which enlarge the entire lymphoreticular system. This occurs as a result of abnormal bone growth crowding the marrow elements from their normal intraosseous position. However, hematopoietic cells are found in irregularly enlarged haversian canals. Frontal bossing is prominent, and the head size is usually large compared with the body. The affected children grow poorly and usually show some degree of mental retardation. Decreased hearing and facial pain are results of nerve involvement, while genu valgum and pectus deformities are common skeletal anomalies. Increased cerebrospinal fluid pressure may occur secondary to bony disease of the calvarium. One of the most serious and common complications is a chronic fistulizing osteomyelitis which .proves resistant to antibiotic therapy and results in septic sequestration of bone. The infection is often compounded by a pancytopenia, and a hemolytic component has been reported.
The dominant or adult form presents as a considerably more benign condition and is often recognized when a routine radiologic examination is performed. Bone pain is another common presenting symptom and is particularly likely to be felt in the lumbosacral region. Fractures, especially of the long bones, occur frequently and heal rapidly, with abundant callus formation. Poor dentition with multiple caries is another common finding, and when teeth are pulled a chronic osteomyelitis of the jaw may develop. Although infections of any bone are common in osteopetrosis, the jaw is an especially characteristic site. Finally, cranial nerve palsies, especially of the third, fourth, and seventh nerves, may be the earliest signs. The disease in adults is very slowly progressive and should be regarded conservatively.
Signs and symptoms referable to the eyes are principally a result of bony abnormalities about the eyes and orbit. Ophthalmoplegia, especially of the third and fourth nerves, as well as nystagmus of inconsistent type, is quite commonly observed. Exophthalmos occurs as a result of bone disease in the orbit and, when extreme, may result in corneal exposure problems. Optic atrophy is potentially the most serious of the ocular complications. It may be due to direct bony compression of the optic nerve, it may be secondary to chronic papilledema, or it may be due to optic nerve involvement by infection in patients who have had chronic meningitis (usually secondary to chronic osteomyelitis ).
Treatment of the infantile form is generally supportive, and the prognosis for survival is poor. Some authors have reported partial success with the administration of corticosteroids in very young children. The treatment of adults is conservative. Supportive measures are taken when fractures or infection occurs.
Fibrous Dysplasia of Bone and Albright's Disease
In 1937 Albright described a syndrome of precocious puberty, dermal pigmentation, and a bizarre dysplasia occurring throughout the skeleton. Since that time, many other cases of histologically similar bone changes have been described; occasionally these are so localized that they have been given the name monostotic fibrous dysplasia. The disease usually begins in childhood, and the presenting signs are referable to the bony anomaly. There is no sex predilection, and no distinct pattern of inheritance has been described. Although not common, it has been described in most parts of the world and should be encountered every few years in a large clinical center (59–61).
Four patterns of bone disease have been described and provide a useful clinical classification of fibrous dysplasia. In the first form, only the orbit is involved, and the symptoms are primarily local. Other bones (e.g., long bones, skull) are also abnormal in the second category. The third form is characterized by involvement of the skull which is so severe that, in effect, an intracranial mass is produced. Finally, the full triad of bone abnormalities, sexual precocity, and skin pigmentation is known as Albright's disease. These four classifications represent. an arbitrary separation of a spectrum of disease. However, since each of these does represent a common clinical presentation, the groupings remain useful.
The bone disease begins in childhood and is usually self-limiting. Termination is common at two times in life: either at or near puberty (with closure of the epiphyses) or in the third decade at about the time that full skeletal maturity is reached. Why in some patients the disease does not progress beyond puberty and in others it goes on to the later date is uncertain. Pathologically, the lesion consists of multiple small osteoid trabeculae surrounded by a vast area of fibrous tissue. Areas of bone are sclerotic and may become much harder than normal. A common presentation in young children is a femoral fracture which occurs after trauma. Often this trauma is minimal, but purely spontaneous fractures do not occur. Healing takes place, but in many cases only after gross disfigurement. Shortened rotated legs and malformed hips are common sequelae. When the lesion affects the bones of the skull and face, fractures are relatively uncommon but disfigurement may be severe. Involvement of the base of the skull (and orbit) tends to occur as a result of maxillosphenoethmoidofrontal bone disease. Although it is often only one bone which is involved, the process may extend between contiguous bones (notably the sphenoid and the frontal). When the skull bone deformity extends intracranially instead of into the orbit or onto the face, neurosurgical intervention is indicated.
Endocrinologic factors have been strongly implicated in the florid syndrome of Albright's disease. Skeletal precocity with early epiphyseal closure has been frequently described. Sexual precocity with menses starting as early as 41/2 years of age and the appearance of secondary sex characteristics as early as 6 years of age is also a well-recognized part of Albright's syndrome. When the early sexual development does occur, the patient is more likely to be female. Occasionally, there is thyromegaly. Several patients with acromegaloid features and 1 patient with gynecomastia have been described.
The pigmented lesions seen in Albright's syndrome are clinically similar to the café au lait spots seen in neurofibromatosis. The lesions may be present at birth and vary in size from millimeters to centimeters in diameter. They are light brown, completely flat, and have the texture of the normal adjacent skin. Pathologically, the lesions consist of areas of increased melanin content in the basal layer of the epidermis. The most common sites of these lesions are the lumbosacral area of the back, the buttocks, and the thighs. Nonocular neurologic deficits include loss of smell, deafness, and any other sign localizing to an area of brain compressed by a mass lesion arising from the bony calvarium.
The eye signs are of three general classes which are all referable to changes in the bony orbit. The most potentially serious change is loss of vision due to compression of the optic nerve within the optic canal. This is easily diagnosed and localized with radiologic techniques; it occurs when the sphenoid bone is involved. The second category of ocular anomaly is proptosis, which, like decreased vision, may be the presenting sign of fibrous dysplasia. The direction of the proptosis is dependent on which of the orbital bones is involved and is usually in a direction away from the mass. Since the tumors may arise from any bone, including the sides of the orbit, the displacement of the globe may be lateral as well as forward. This lateral displacement of the globe would be a very unusual finding in the majority of other orbital tumors. The sequelae of proptosis, such as corneal exposure and chemosis, are found when the displacement is severe. The final common ocular finding is a disturbance in motility, which occurs for obvious reasons. It should be emphasized that the monostotic form of the disease often shows a predilection for the bones of the orbit. The absence of generalized findings should in no way dissuade the clinician from the diagnosis of fibrous dysplasia.
The disease is self-limited, and treatment is basically supportive. When the optic nerve is compressed or an intracranial mass lesion is present, decompressive surgery must be undertaken. Fractures should be set promptly and cosmetic surgery on the skull and face undertaken when necessary. When the indications for orbital surgery are minimal, such therapy is often unrewarding. Surgery for strabismus should also be approached cautiously and only when the disease is quiescent.
Conradi's Syndrome
Conradi's syndrome (chondrodystrophia calcificans congenita, C.C.C., chondrodysplasia epiphysialis punctata, the stippled epiphyses syndrome) was first described by Conradi in 1914. It is a rare disease, the precise incidence of which cannot as yet be established. The disease has been reported in both blacks and whites and from all parts of the world. Although early reports indicated that the disease occurs in a ratio of two to three females per male, more recent findings indicate the sex ratio to be much closer to unity. The disease has an autosomal recessive inheritance and a high degree of penetrance. It is usually described as having eight characteristic groups of features. These include contractures and micromelia of the extremities, various dermatoses of the skin, characteristic eye findings, saddle nose deformity, positive familial history, and, finally, the pathognomonic radiologic findings in the epiphyseal regions of the bones (62–66). In general, the prognosis for survival in these children is poor: death occurs within 5 months in 50% of patients. Mental retardation is usually present.
Bone changes are highly typical and involve much of the skeleton. The stature is short, and linear skeletal growth is below normal. Kyphosis and scoliosis are common vertebral anomalies, and micromelia and contractures occur in the extremities. Although the lower extremities are usually more severely affected than the upper extremities, both the humerus and the femur are shortened, with epiphyseal and extraepiphyseal calcification. Radiologically these calcifications take the appearance of snowflakes in the area of the epiphyses and are quite symmetric. This finding may regress by 2 to 3 years of age. Bilateral congenital hip dislocation and various hand and foot deformities, such as syndactyly and polydactyly, have been reported. Several patients have shown clubfoot deformity.
Craniofacial anomalies are quite common; various craniostenoses, macrocephaly or microcephaly, and frontal bossing have been reported. The face shows a typical wide and depressed nasal root. Some of the small nasal bones are occasionally absent. In the mouth a high arched palate may be seen along with multiple dental anomalies. The skin and scalp are usually abnormal: dry, scaly, and atrophic patches (atrophoderma follicularis) are frequently seen. There is loss of scalp hair due to cicatricial alopecia, and the remaining hair is coarse. Other skin changes, such as seborrheic dermatitis and ichthyosis, have been described.
Visceral changes, although not comprising the major part of the syndrome, are occasionally seen. The most common are cardiac anomalies, including patent ductus arteriosus and ventricular septal defects. Other systemic findings are extensive fibrous tissue replacement of muscles, a mucoid degeneration and calcification of the connective tissue, and umbilical and inguinal hernias.
Ocular anomalies form an integral part of Conradi's syndrome. Externally, hypertelorism is present in nearly all patients, and sparse hair of the eyebrows is irregularly evident. Most characteristic are bilateral dense cataracts, which are usually congenital but may develop in youth. Occasionally, there is a unilateral cataract at birth and a cataract develops in the other eye at some future date. A general feature is that patients with congenital cataracts tend to have a poorer prognosis than those without them. The skin changes are associated with the cataract. One case has been described in which glaucoma and an anomalous chamber angle along with heterochromia and hypoplasia of the iris were present. Another common ocular anomaly is bilateral severe optic atrophy, the exact causes of which have not been determined.
Treatment is usually unrewarding. Since most patients die in infancy or early childhood of intercurrent infection, surgical treatment aimed at correcting skeletal anomalies should be postponed until a time in childhood (about 4 years old) when survival is more secure. The same philosophy should be applied to the treatment of cardiac disorders, unless they are life-threatening. The prognosis for successful cataract surgery is poor, not only because of the often unfavorable results of surgery in congenital cataracts but also because of the frequent presence of optic atrophy.