The synthesis of thyroid hormones is dependent on the availability of dietary iodine, which is incorporated by the thyroid gland into individual tyrosyl residues of a complex molecule called thyroglobulin. These individual residues merge to form T3 and T4, which are cleaved from the parent thyroglobulin molecule for release into the blood stream. Thyroid hormones are transported in the blood stream bound principally to thyroxine-binding globulin. Only a very small percentage (less than 0.5%) circulate “free” or unbound, and it is this fraction that is responsible for the physiologic action.
The function of the thyroid gland is controlled by the anterior pituitary gland via the secretion of thyroid-stimulating hormone (TSH), which is itself influenced by the release of thyrotropin-releasing hormone by the hypothalamus; TSH release by the anterior pituitary is partly controlled by the thyroid hormones through a negative feedback mechanism.
HYPERTHYROIDISM
Hyperthyroidism or thyrotoxicosis is characterized by the overproduction of thyroid hormones. A small proportion of patients have only elevated T3 levels, but the majority possess high levels of both T3 and T4. Thyrotoxicosis may be classified broadly into two groups: (1) where the thyroid gland is either diffusely hypertrophic and hyperplastic (Graves' disease); or (2) where single or multiple hyperactive nodules exist in the gland.
Graves' disease is a condition that predominantly affects females and, although it may occur at any age, has a peak incidence in the third and fourth decades. In almost all patients with this autoimmune disease, immunoglobulins are directed against the TSH receptors on the thyroid cellular membrane. As with other autoimmune conditions, there is a strong familial tendency for Graves' disease: it has been associated with the HLA antigens B8 and DR3 in whites and with Bw35 in Asians.
It usually presents insidiously, although rapid and dramatic presentations are not uncommon; the exact precipitating mechanism is unknown. Systemic symptoms include weight loss, increased sweating and heat intolerance, palpitation, fatigue, and diarrhea. Examination of a patient with Graves' disease may reveal a goiter, a fine tremor, palmar erythema, finger clubbing, vitiligo, alopecia, pretibial myxedema, and a multitude of cardiovascular signs (e.g., tachycardia, atrial fibrillation, bounding peripheral pulses).
Ocular Involvement
Ocular involvement is an almost integral part of the clinical presentation of Graves' disease; however, the systemic and eye manifestations are generally considered to run independent courses. Although most of the systemic features of Graves' disease can be attributed to the increased activity of thyroid hormones, the ocular involvement is less obviously linked to this. Graves' ophthalmopathy, preferably termed thyroid-associated ophthalmopathy (TAO), may occur in the absence of overt systemic signs of hyperthyroidism (approximately 10% of cases; more subtle analysis often reveals evidence of systemic thyroid disease in most of these cases), or even after the systemic disease has been treated adequately. TAO is the most common cause of proptosis in adults; the term exophthalmos is used exclusively to describe the proptosis of TAO and will be used henceforth in this section.
Compared with Graves' disease, hyperthyroidism due to “toxic” thyroid nodules is less common and generally occurs in older patients. Ocular involvement is not a common feature of this condition.
Presentation
Patients with TAO may present with a wide range of symptoms. Both eyes are usually affected, often to differing degrees, or the involvement may be unilateral. Although the condition may be selflimiting, it often has a tendency to produce episodes of acute activity lasting several months interspersed by relative quiescence, but the progression in severity from any one acute episode is not likely to regress as a result of fibrotic changes. The natural tendency of the disease process is to eventually “burn out” within 2 to 3 years.
Lid puffiness and irritable, watery eyes are common symptoms; complaints include a feeling of “grittiness” in the eyes and profuse tearing despite the absence of apparent irritants. Affected eyes may feel gritty for several reasons: (1) lid retraction and exophthalmos may cause problems with corneal lubrication, and superior limbic keratitis is a well-recognized association; (2) thickened conjunctival folds may obstruct the drainage of tears into the lower lid punctum, evidenced by a fullness of the conjunctiva at the medial canthus; and (3) conjunctival hyperemia commonly accompanies inflammation of the extraocular muscles. The injection is usually most marked over the insertions of the extraocular muscles (Fig. 1).
|
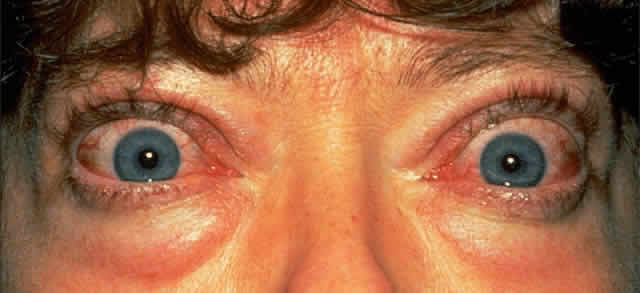
Cosmetic problems associated with protruding eyes are also common; this protrusion is usually asymmetric and is due to a combination of upper lid retraction and exophthalmos. Lid retraction is among the earliest and most common signs causing exposure of the superior sclera and widening of the palpebral fissure. It is one feature of TAO that can be partly attributed to the systemic effects of hyperthyroidism; increased stimulation of the smooth muscle retractors in both the upper and lower lids can result from heightened adrenergic activity. Upper lid retraction also results from a tightened inferior rectus due to inflammation and fibrosis, and this tightening leads to compensatory overactivity of the superior rectus/levator palpebrae complex. Similar inflammatory and fibrotic involvement of the levator palpebrae muscle will add further to upper lid retraction.1 This wide-eyed staring appearance is often accentuated by the presence of exophthalmos (see Fig. 1).
When mild, the signs and symptoms of exophthalmos may be limited to a feeling of grittiness, conjunctival injection, and chemosis; however, severe corneal exposure, which is often complicated by a poor Bell's phenomenon because of restrictive myopathy, can lead to pannus formation and ulceration, corneal scarring and infection, and visual impairment. Because exophthalmos is secondary to increased retro-orbital mass, it can be associated with optic nerve compression, although significant nerve compression may be present in its absence.
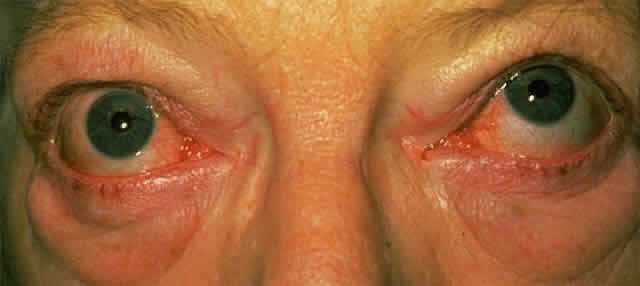
The principal visual symptoms are diplopia and reduced visual acuity. According to Fells,2 patients with thyroid-associated diplopia see vertically separated images upon waking in the morning, which improve within minutes but recur later during periods of fatigue or alcohol intoxication. Defective ocular motility in TAO is due to inflammatory engorgement of the extraocular muscles: the inferior and medial recti more commonly and the lateral and superior recti less commonly. The restriction in function may initially be reversible, but is eventually permanent due to cicatricial, fibrotic elements (Fig. 2). Since the inferior rectus muscle is the most commonly affected, upgaze can often lead to elevated intraocular pressure.
|
Optic nerve compression (compressive neuropathy) is one of the serious complications of TAO and leads to reduced visual acuity. Its prevalence has been reported at approximately 10% in a prospective study of 101 patients attending a combined thyroid-eye clinic during a 5-year period3; this figure may be artificially high because of the specialized nature of this clinic. It is more likely that less than 5% of patients with Graves' disease will develop sight-threatening compressive neuropathy. Optic nerve involvement is generally accepted to be due to compression at the orbital apex by enlarged extraocular muscles.4 These patients are likely to be older and male; they commonly exhibit limitations of extraocular movements with significant vertical deviation.5,6 Other causes of reduced visual acuity are corneal exposure and desiccation, with their attendant complications, and chorioretinal striae involving the macula, due to an increased retro-orbital mass.
In an attempt to clarify the nomenclature pertaining to thyroid-related eye disease, Werner classified the eye changes into seven categories, based on an original classification by the American Thyroid Association7; Werner's classification is presented in Table 1. Although useful as a means of classifying the severity of involvement, its usage is limited because it lacks a means of measuring the rate of progression or treatment-induced regression of clinical activity. The signs of TAO can be subdivided into those pertaining to the lids, conjunctiva and cornea, globe, extraocular muscles, and fundus. These signs are summarized in Table 2.
TABLE 1. Werner's Classification of Thyroid-Associated Ophthalmopathy*
| Class | Definition |
| 0 | No signs or symptoms |
| 1 | Only signs, no symptoms (signs limited to upper lid retraction ± lid lag and proptosis) |
| Proptosis is graded into: | |
| 0: Absent (20 mm or less) | |
| a: Minimal (21–23 mm) | |
| b: Moderate (24–27 mm) | |
| c: Marked (28 mm or greater) | |
| 2 | Soft tissue involvement |
| i: Symptoms of excessive lacrimation, “gritty” sensation, retrobulbar discomfort, and photophobia | |
| ii: Signs are subdivided into the following grades: | |
| 0: Absent | |
| a: Minimal (edema of conjunctiva and lids, conjunctival injection, orbital fat extrusion, palpable lacrimal glands, swollen extraocular muscles) | |
| b: Moderate (minimal signs plus chemosis, lagophthalmos, eyelid “fullness”) | |
| c: Marked | |
| 3 | Proptosis (subdivided as in class 1) |
| 4 | Extraocular muscle involvement |
| 0: Absent | |
| a: Minimal (limitation of motion, evident at extremes of gaze) | |
| b: Moderate (evident restriction of motion without fixation of position) | |
| c: Marked (fixation of position of a globe or globes) | |
| 5 | Corneal involvement |
| 0: Absent | |
| a: Minimal (stippling of cornea) | |
| b: Moderate (ulceration) | |
| c: Marked (clouding, necrosis, perforation) | |
| 6 | Sight loss (optic nerve compression) |
| 0: Absent | |
| a: Minimal (disc pallor or choking, or visual field defect; vision 20/20–20/60) | |
| b: Moderate (minimal signs, but vision 20/70–20/200) | |
| c: Marked (vision less than 20/200) |
* As indicated, each class is further subdivided into absent, mild, moderate, or marked. The first two classes (0 and 1) represent non-sight-threatening disease, the latter five classes (2 to 5) represent more severe infiltrative disease, increasing numbers signifying more advanced involvement. The first letters of each definition constitute the acronym NO SPECS, which is the name by which this classification is commonly referred.7
(Adapted from Werner SC: Classification of the eye changes of Graves' disease. Am J Ophthalmol 68:646, 1969.)
TABLE 2. Signs of Thyrotoxicosis
Lids
Puffy lids (Enroth's sign)
Lid retraction (Dalrymple's sign)
Lid lag:
- Delay of upper lid in following globe movement in downward gaze (Graefe's
sign)
- Jerky downward movement of the eyelid (Boston's sign)
- Delay of lower lid in following globe movement in upward gaze (Griffith's
sign)
- Globe lagging behind upper lid on upward gaze (Mean's sign)
Tremor of closed eyelids (Rosenbach's sign)
Reduced blinking (Stellwag's sign)
Increased pigmentation of skin of the eyelids (Jellinek's sign)
Conjunctiva and Cornea
Injected, chemotic conjunctiva
Superior limbic keratoconjunctivitis
Signs of corneal exposure ± pannus formation and ulceration
Globe
Exophthalmos
Ocular bruit
Extraocular Muscles
Apparent overactivity of extraocular muscles, most commonly:
- Inferior rectus with weakness of upgaze
- Medial rectus with weakness of convergence
Elevated intraocular pressure on upgaze
Fundus
Congested retinal veins
Swollen optic disc
Chorioretinal striae
Pale, atrophic optic disc
Pathogenesis
Present understanding of the pathogenesis of TAO remains unclear; however, as with the thyroid component, autoimmune mechanisms have been implicated. The association between the ocular disorder and a hyperactive thyroid gland may be the result of a linkage between the two conditions and a primary autoimmune disorder manifested through a variety of circulating autoantibodies acting on two different end-organs sharing a hypothetical autoantigen. Currently it is unclear what this autoantigen is, but several candidates have been proposed, including a 64-kilodalton (kd) protein found in thyroid and eye-muscle plasma membranes8,9 and the TSHreceptor protein.10,11 Antibodies directed against a 64-kd protein expressed by extraocular muscle has been reported to be present in 33% of patients with TAO and in 75% of patients with severe eye disease.12 There may also be other antigens expressed in the orbit similar to both thyroglobin and antigens expressed on thyroid microsomal cells, since antibodies directed against them have been identified in the orbit.13
The stimulatory strength of these respective antibodies vary among patients. In cases predominantly involving the eye, the antibodies directed against the thyroid may be weak. In cases where these antibodies prevent the normal binding of TSH, there may be an association between hypothyroidism and TAO. It has also been suggested that a common autoantigen exists (e.g., the 64-kd antigen) that is responsible for the milder eye disease seen in the majority of patients with Graves' disease; in patients with severe disease, other eye-muscle-specific antigens have been proposed.14 It has been demonstrated that IgG from patients with TAO stimulated the growth of extraocular myoblasts, compared with sera from controls and patients with Graves' disease who had no ocular involvement. The effect was relatively specific to these cells, whereas the effect on skeletal myoblasts was less marked.15
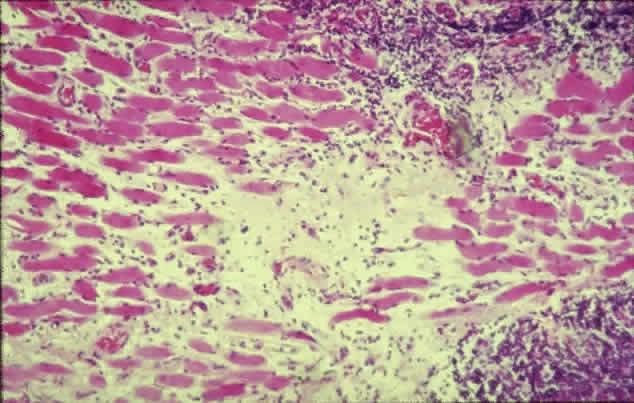
The inflammatory infiltration of the extraocular muscles consists principally of activated T-cells together with smaller numbers of B-cells, macrophages, and mast cells.16 The infiltration is mainly interstitial and is accompanied by increased fibroblastic activity induced by cytokines derived from the immune cells, leading to deposition of glycosaminoglycans and collagen as well as edema. The eventual outcome is fat cell infiltration and fibrosis (Fig. 3). The muscle fibers appear normal, with the exception of subsarcolemmal deposits of lipid and glycogen, and there is an absence of muscle-cell destruction.
|
It is unclear which cell type expresses the offending antigen, but current understanding favors the fibroblast. According to Weetman,17 the histologic picture reveals a pathologic process directed principally against the retrobulbar fibroblast, rather than muscle cells, but this may be just a manifestation of the presence of large numbers of immune cells attracted there by a nonfibroblastic source. Antibodies against the TSH-receptor have been shown to stimulate collagen synthesis by fibroblasts18; retrobulbar fibroblasts have also been demonstrated to possess TSH-receptor-encoding RNA.19 However, an immune process directed against muscle cells is suggested by the finding of higher levels of eye-muscle-binding antibody in patients with TAO, whereas levels of antifibroblast antibody were unremarkable.20
Also unclear is why the extraocular muscles are selectively involved in TAO while other skeletal muscles in the body are spared. Schmidt and associates21 reported that hyperthyroidism alters the immunocompetent cell population in extraocular muscles, but not skeletal muscles. Another possible explanation is that extraocular muscles also possess more spindles and connective tissue and have a greater blood supply than skeletal muscles.
Most of the infiltrative pathology seen in TAO traditionally has been attributed to the retro-orbital deposition of glycosaminoglyans, with associated edema and inflammatory cell infiltration of the muscles. More recently, Hudson and colleagues22 proposed that, in some cases, the exophthalmos may be due to passive orbital venous congestion from a partial obstruction of the superior ophthalmic vein. They based this view on the observation that exophthalmos is sometimes present with little enlargement of the extraocular muscles, with the exception of the superior rectus muscle together with congestion of the superior ophthalmic vein with which it is closely linked. The obstruction could be purely compressive, or there may be an element of contiguous inflammation from the adjacent muscle.
Orbital fat content is not thought to play a significant role in compressive optic neuropathy. Feldon and co-workers23 demonstrated that patients with optic nerve compression had greater extraocular muscle volumes than those without optic nerve involvement, whereas both groups had similar fat content; orbital fat volume was found to decrease with increasing muscle size.
Diagnosis
Diagnosis of TAO is based on clinical findings, sometimes in the absence of hyperthyroidism. Symptoms and signs indicating active inflammation (e.g., pain, conjunctival injection, edema) are particularly important because they influence the management strategy.24 The degree of exophthalmos should be assessed with an exophthalmometer (a protrusion of 20 mm or more is suspicious), and the intraocular pressure should be measured with the patient in the primary position and with attempted upgaze. Photography is a useful method of recording clinical status.
Clinical investigations may reveal elevated plasma T3 and T4 levels (the free thyroxine index can be determined after correction for plasma protein binding) and in most cases decreased plasma TSH. Antibodies that may be detected include those directed against TSH-receptor, thyroglobulin, and thyroid membranes.
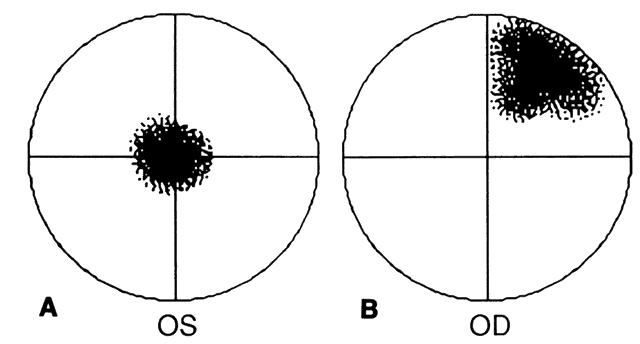
Computed tomography (CT), most revealing in the coronal plane, and ultrasonography are useful methods for assessing extraocular muscle thickness (Fig. 4). Magnetic resonance imaging (MRI) has been advocated as a means of differentiating between extraocular muscles that are actively inflamed and those that are fibrosed25–27; both muscular problems can restrict ocular movement, but the former is amenable to immunosuppressive intervention. The T2 relaxation time of MRI is directly proportional to the water content of the tissue scanned: since inflamed tissues are edematous, they should have longer T2 times. MRI is also thought to be better than CT in providing views of the orbital apex and may therefore be better for evaluating optic nerve compression.2
|
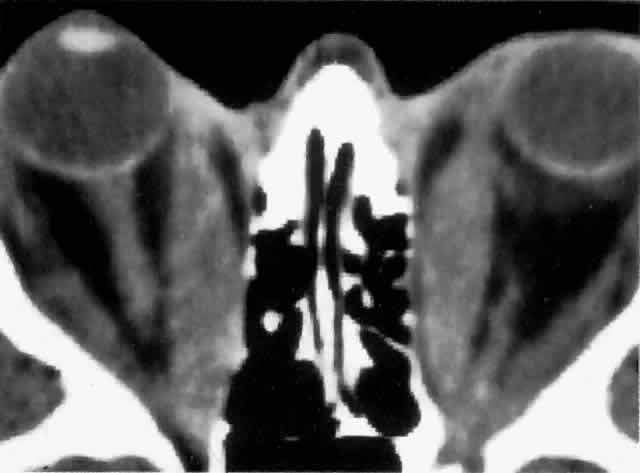
Extraocular muscle volume is inversely proportional to ocular motility, especially for horizontal movements. Therefore, the peak velocity of saccadic eye movement, measured with reflective infrared oculography, has been proposed as another means of assessing clinical severity.28–30 The degree of optic nerve compression has been found to increase as peak velocity decreases, particularly with larger angles of eye movement.29
Patients with optic nerve compression present with reduced visual acuity, which may be clinically substantiated by the presence of an afferent pupillary defect and visual field loss. More sophisticated means of detecting early compressive pathology include the pattern electroretinogram, cortical visual evoked potentials, and color contrast sensitivity along the tritan axis.31 Depending on the degree of severity, fundal examination may reveal varying degrees of venous congestion, swollen optic discs, and chorioretinal striae (usually horizontal). It is important to note that 40% to 50% of patients with optic nerve compression may have a normal fundal appearance.5,32
Prognosis and Risk Factors
Unlike Graves' disease, which is far more prevalent in females, TAO is much more evenly distributed between the sexes; in fact, when TAO is present in males, there is a greater likelihood of more severe involvement.3 TAO is a part of Graves disease and therefore cannot be differentiated from it reliably. Older patients are more likely to develop more severe ocular disease, possibly because of age-dependent alterations in the elasticity of the extraocular muscles, rendering them stiffer and more likely to impinge on the optic nerve rather than molding around it.29 Kendler and associates,33 in a series of 557 cases of Graves' disease, reported that patients older than 50 years have an increased incidence of ocular motility impairment (32% versus 12%) and more severe reduction in visual acuity.
Smoking is strongly associated with an increased risk of TAO development as well as with more severe involvement34–36; this effect may be related partly to a smoking-induced increase in thyroglobulin release from the thyroid gland and to alterations in immunoregulatory cell function.
Various forms of treatment for hyperthyroidism, including radioactive iodine37,38 and surgery,39 have been found to increase the severity of TAO. It has been suggested that this may be due to the increased release of antigenic thyroidal elements into the blood stream. The presence of pretibial myxedema is a poor prognostic sign and usually signifies the presence of significant ophthalmopathy and a prolonged clinical course.
Management
It is unclear whether treatment of the underlying thyrotoxic state produces any reduction in the ocular symptoms apart from lid retraction, which if it is secondary to increased adrenergic activity, may be expected to improve. If the cornea is exposed, it is important to prescribe artificial tears as a means of corneal lubrication. To help reduce the degree of periorbital edema accumulation overnight, the patient should be instructed to sleep on several pillows or to have the mattress at the head end of the bed elevated.
For more severely affected eyes, immunosuppressive therapy with glucocorticoids and retrobulbar irradiation has been reported to benefit approximately 60% of patients with TAO, particularly in improving visual acuity and cosmesis; this treatment is less effective in combating exophthalmos and movement dysfunction.40 Immunosuppressive agents have the greatest effect against active inflammatory disease of short duration, rather than changes that are principally cicatricial. Plasmapheresis has also been used to reduce the degree of immune activity, although it usually has only a transitory effect.
Steroids are the most widely used immunosuppressive drugs; to be effective, high doses of prednisolone, 80 to 100 mg daily, are usually required and relatively high doses are often needed during a course of several weeks to maintain the effect. The addition of a second drug, such as cyclosporin A or azathioprine, may allow for lower doses of prednisolone. Based on the fact that methylprednisolone has been used with good effect in other autoimmune conditions, Kendall-Taylor and colleagues41 described the successful use of a large-dose IV bolus of methylprednisolone, carefully monitoring the side effects, particularly those involving the cardiovascular system. Their protocol used a dose of 0.5 g methylprednisolone in 200 mL isotonic saline infused in a 30-minute period, with a second 0.5 g given 48 hours later. A maintenance dosage of oral prednisolone, 40 mg daily, was then commenced; within a four-week period, it is gradually reduced to 10 mg. Subsequent reduction in dosage depended on clinical status. The principal advantages of this technique over entirely oral prednisolone appears to be the rapid improvement in visual acuity (within 48 hours) and a relatively lower incidence of side effects.
Low-dose external-beam megavoltage radiotherapy has been employed successfully in treating sight-threatening TAO; the improvement in radiotherapy techniques, principally the use of tightly collimated beams with small treatment portals through a lateral port, means that the incidence of long-term side effects such as cataract formation and radiation retinopathy is low.42 A total dose of 20 Gy delivered in 12 fractions in a period of 15 days is a recommended treatment regimen.43
A prospective study by Prummel and co-workers reported similar efficacy between steroid therapy and radiotherapy in patients with moderately severe TAO, but without optic nerve involvement (approximately 50% of patients responded in each group); of patients treated with steroids, a higher proportion had side effects compared with those treated with radiotherapy.44 Prednisone (a 3-month oral course) was found to be more effective in reducing soft tissue edema, whereas radiotherapy (2 Gy daily in 10 fractions in a period of 2 weeks) was considered better at improving motility. Because of the lack of patients with compressive optic neuropathy, the primary use of radiotherapy in these patients currently cannot be advocated because of its delayed effect.45
If medical decompression is not successful (i.e., vision remains poor, continues to deteriorate or corneal exposure remains severe), then surgical decompression is the next treatment option. Decompression allows some of the congested orbital contents to prolapse into the surrounding sinuses. With improvements in surgical technique, surgical decompression is also frequently performed in patients with lesser degrees of involvement to improve cosmesis.46 Several approaches toward this end have been described, ranging from the minimum requirement of removing the medial wall and medial half of the orbital floor, to four-wall decompression; although the latter achieves the greatest degree of retrodisplacement, it is also associated with the highest complication rate and therefore is not commonly performed.
Orbital decompression in cases of compressive neuropathy has been reported to have high success rates, approximately 90% of patients experiencing an improvement in vision or maintaining visual stability; no major complications have been reported after transantral decompression.47,48 From the results of a postal survey of ophthalmic surgeons, McCord49 reported that 41% of orbital decompressions were performed to reduce corneal exposure from severe exophthalmos, 39% to alleviate optic nerve compression, and 20% to improve the cosmetic impairment caused by milder forms of exophthalmos. The series also described antralethmoidal and three-wall decompressions (medial, floor, and lateral walls) as producing an average of 4 to 6 mm of retrodisplacement of the globe; the antral-ethmoidal approach was the most commonly applied (75% of the surgeons surveyed). Decompression may be associated with worsening extraocular muscle imbalance postoperatively. The translid approach is thought to produce fewer problems than the transantral approach, although the latter is more efficacious in relieving nerve compression because it allows decompression up to the orbital apex. Orbital decompression for lesser degrees of exophthalmos with removal of orbital fat without bone also has been described, and a retrodisplacement of 2.2 to 5.9 mm has been achieved.50,51
Up to 15% of patients with TAO may suffer from diplopia.52 Regular orthoptic assessment is needed to manage this symptom satisfactorily. Diplopia may be relieved initially with the application of temporary prisms (e.g., Fresnel prisms), the adoption of a compensatory head posture, or in severe cases, occlusion. After a period of stability (Fells2 recommends a period of 6 months), extraocular muscle surgery, or in milder cases, the incorporation of prisms into the spectacle prescription, may be contemplated. The most common form of extraocular muscle surgery is the recession of tight, fibrous inferior rectus muscles with the aid of adjustable sutures to optimize positioning.
The use of botulinum toxin in patients with TAO suffering from diplopia has been advocated by some.53,54 Lyons and associates53 injected 62.5 picograms of botulinum neurotoxin A in 0.1 mL normal saline into the affected muscle under electromyographic monitoring in a series of 38 patients with an average hypotropia of 23 prism diopters and esotropia of 28 prism diopters; 75% of the patients achieved a mean reduction of 14 prism diopters in the angle of deviation. The effect was undetectable after 2 months in the majority of patients, most of whom eventually required strabismus surgery. Six patients did obtain long-term stability (at least 12 months); however, of these patients three had previously undergone strabismus surgery. Lyons and colleagues advocated this form of treatment for two reasons:
- With conventional treatment, patients with diplopia must wait for the condition
to stabilize before surgery can be contemplated, which may mean
that they will have a prolonged period of visual disturbance. There
is one caveat, however: botulinum toxin will interfere with orthoptic
assessments during this period.
- There is a possibility that the improvement achieved with botulinum toxin
will be permanent.
Lid retraction may be improved by orbital decompression, especially of the lower lid; however, the backward and downward movement of the globe following decompression may accentuate upper lid retraction. Repositioning (recession) of the upper lid retractors may have to be performed as an adjunct. To improve cosmesis in patients who do not have significant exophthalmos, primary recession of the lid retractors can be performed without decompression. Any required lid procedures, however, should be undertaken after decompression (which may worsen extraocular muscle imbalance) and strabismus surgery. Such techniques include disinsertion of Müller's muscle, with or without levator aponeurosis incision or recession for upper lid retraction. Botulinum toxin has also been used to reduce lid retraction, producing an improvement lasting up to 32 weeks.55 Spacers may be inserted between the lower lid retractors and the tarsal plate to improve lower lid retraction; sclera and hard palate mucosal grafts have been employed to this end.
HYPOTHYROIDISM
Most commonly, hypothyroidism occurs secondary to idiopathic atrophy of the thyroid gland; it may also result from treatment for hyperthyroidism (particularly after radioactive iodine therapy), iodine deficiency, and Hashimoto's thyroiditis. Hypothyroidism usually has an insidious onset: patients present with complaints of lethargy, weight gain, dry and thickened skin, coarse hair, anorexia, constipation, thickening of the voice, and psychiatric symptoms.
Ophthalmologic features of the condition include periorbital swelling, which is part of the generalized nonpitting skin edema of myxedema and the characteristic loss of the outer third of the eyebrows. A more unusual ophthalmic feature is open-angle glaucoma, which has been reported to be associated with the deposition of a mucopolysaccharide within the trabecular meshwork.56