NEVUS
An iris nevus is a benign tumor that arises in the melanocytes in the iris stroma.2 Although most iris nevi remain clinically stationary, they can occasionally give rise to malignant melanoma. Histopathologically, an iris nevus is usually composed of low-grade spindle-type cells. Occasionally it is composed of deeply pigmented, round cells similar to those seen in the melanocytoma of the optic disc.
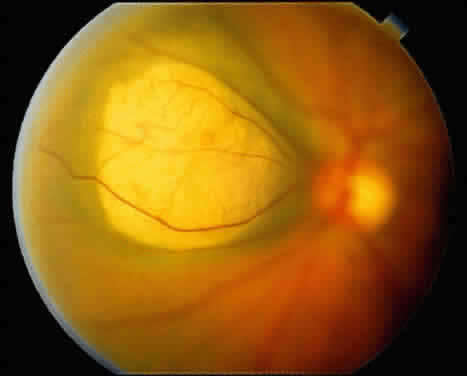
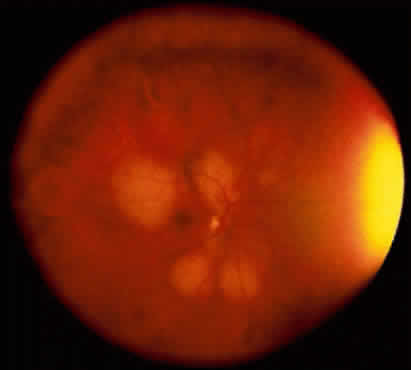
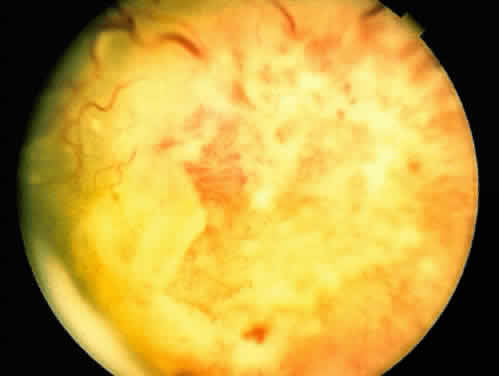
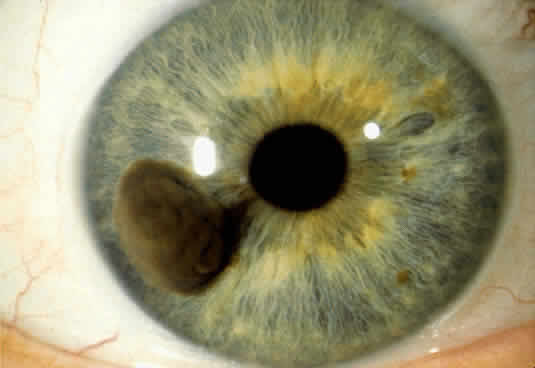
An iris nevus classically appears as a variably pigmented, well-circumscribed lesion in the iris stroma (Fig. 1). It may involve any portion of the iris from the pupillary border to the iris root and may be flat or minimally elevated. Occasionally, an iris nevus may occupy an entire sector of the iris from the pupillary border to the anterior chamber angle. In some cases, an iris nevus may be clinically amelanotic. An iris nevus can induce an irregular pupil or sector cortical cataract.
|
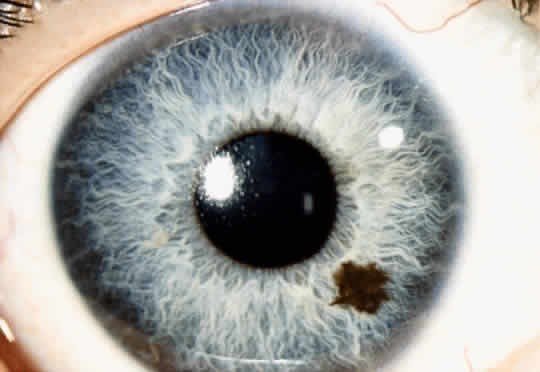
Iris nevus is best diagnosed by recognition of the typical lesion with slit-lamp biomicroscopy. Ancillary diagnostic studies, such as fluorescein angiography and ultrasonography, have little additional diagnostic value. Management includes documentation with accurate drawings or photographs and examination on a yearly basis, looking for evidence of enlargement of the lesion. If growth is detected, malignant transformation into melanoma should be suspected.
MELANOMA
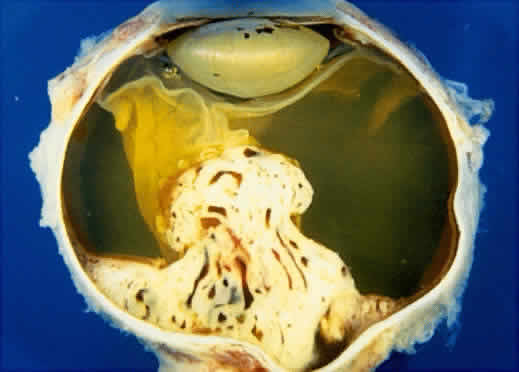
An iris melanoma is a malignant melanocytic tumor that arises from the melanocytes of the iris stroma.2 Histopathologically, it is composed of spindle cells, epithelioid cells, or a combination of the two (mixed-cell type).
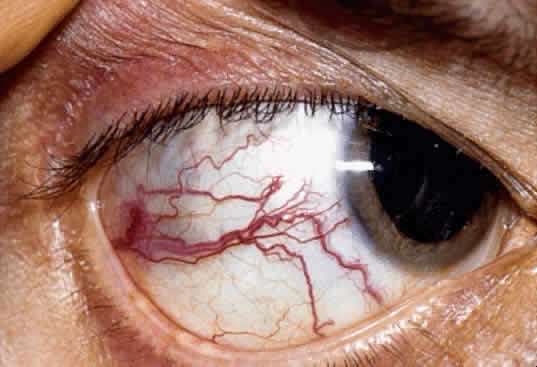
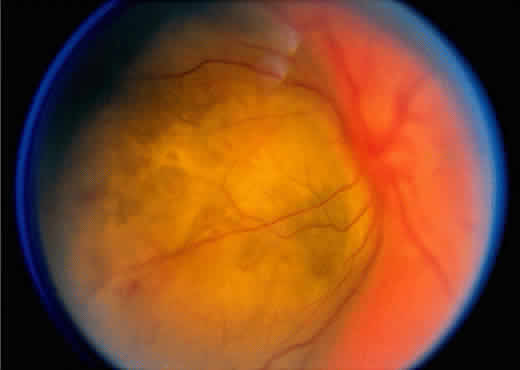
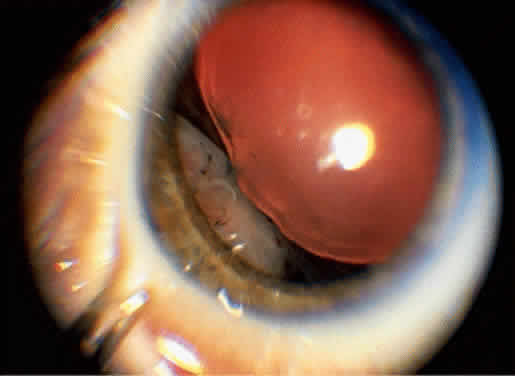
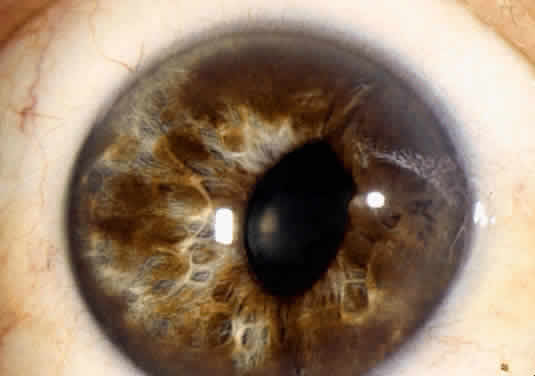
An iris melanoma is characterized clinically as a variably pigmented, elevated, circumscribed or diffuse, melanocytic neoplasm that affects the iris stroma. It is typically larger than an iris nevus. The lesion most often is deeply pigmented and elevated (Fig. 2). In can be amelanotic, or it can be partially pigmented and partially nonpigmented.
|
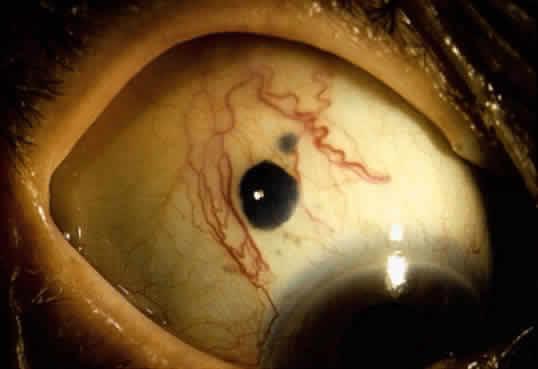
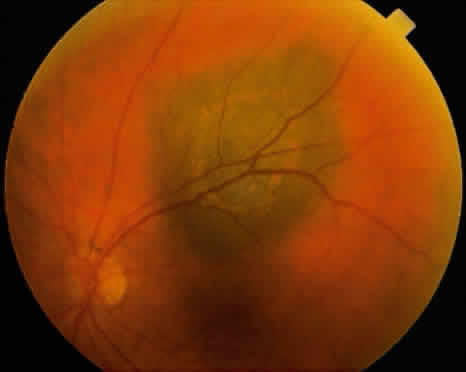
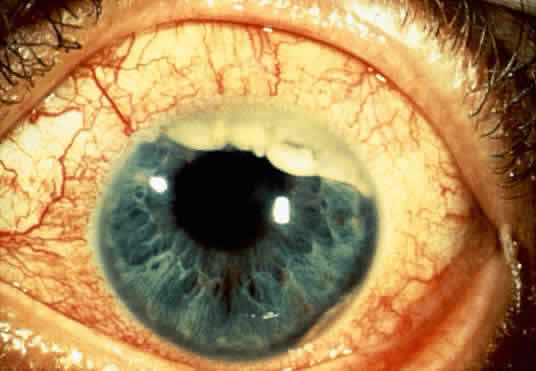
An important variant is the diffuse iris melanoma. It grows as a flat, diffuse, often multifocal mass that covers a large area of the iris surface (Fig. 3). The patient with a diffuse iris melanoma typically presents with a clinical syndrome of progressive acquired hyperchromic heterochromia and ipsilateral secondary glaucoma.
|
Iris melanoma has the capacity to exhibit distant metastasis to the liver and other organs. Lesions that are diffuse and produce secondary glaucoma tend to have a greater tendency to spawn metastatic disease.3
The diagnosis of iris melanoma is best made by an experienced observer who can recognize its typical features on slit-lamp examination. Although fluorescein angiography and ultrasonography have been employed, they add little useful diagnostic information. Fine-needle aspiration biopsy can be employed to confirm the diagnosis of diffuse iris melanoma in cases where enucleation is being considered.4
Once the diagnosis of iris melanoma is clearly established, usually by documentation of progressive growth, the best management is surgical excision of the lesion. Lesions confined to the iris can be managed by removal by partial sector or peripheral iridectomy. Those that extend into the ciliary body require iridocyclectomy or iridogoniocylectomy. The diffuse iris melanoma is often too large to resect locally and may require enucleation or plaque radiotherapy in selected cases.5
ADENOMA OF IRIS PIGMENT EPITHELIUM
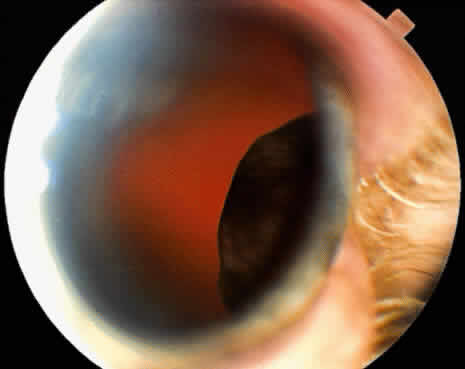
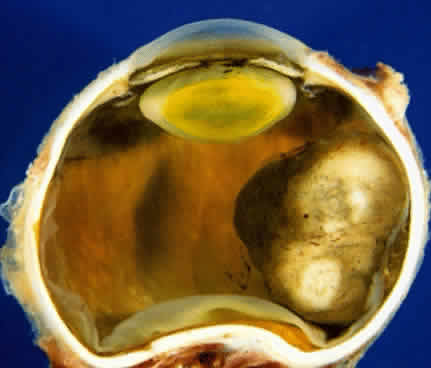
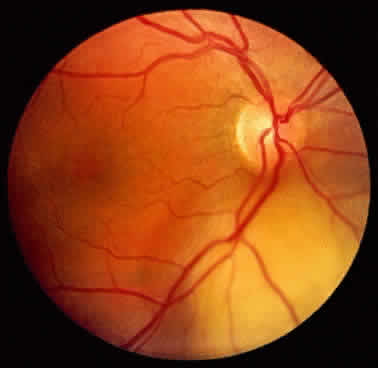
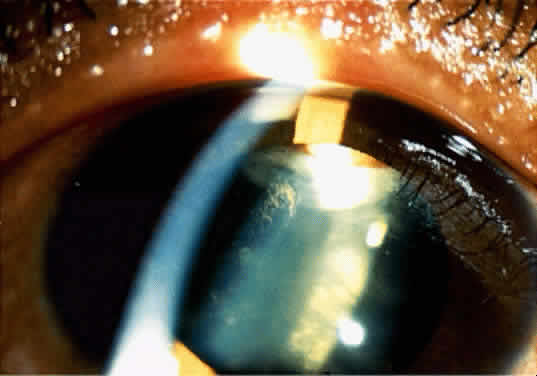
Adenoma of the iris pigment epithelium is a benign neoplasm that arises from the posterior pigment epithelium of the iris.6 Histopathologically, it is composed of columns or acini of pigmented epithelial cells. Clinically, it appears as a dark black lesion, seen in the angle as a rounded mass or as an irregular, multinodular mass (Fig. 4). The lesion typically remains stable or progresses very slowly. Transformation into adenocarcinoma is exceedingly rare.
|
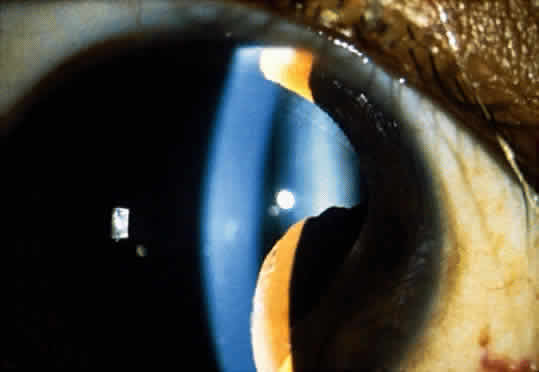
The diagnosis of adenoma of the iris pigment epithelium is best made by clinical recognition of the characteristic features. Small, asymptomatic tumors can be managed by simple observation. If the lesion shows progressive enlargement or early secondary glaucoma, removal by iridectomy or iridocyclectomy is warranted.