CLINICAL FEATURES
Ocular herpes simplex infection is a leading cause of corneal blindness in the developed world. The estimated prevalence is roughly 150 per 100,000 population.4 Experimental infection can be induced in other animals, but humans are the only natural hosts of HSV, and infection is ubiquitous among them. It is estimated that by 15 to 25 years of age, 70% of the population has been infected, and this proportion rises to 97% by 60 years of age.5 Transmission of the virus is by direct contact, and it has an incubation period of 3 to 6 days. The initial infection is subclinical in 85% to 99% of cases, and all infected individuals continue to carry the virus. The oral mucosa is most commonly infected by contaminated saliva droplets, and the virus can be transmitted to the eye by way of the trigeminal nerve. Ocular HSV-2 infection is likely spread directly by oculogenital contact or by contaminated fingers, but hematogenous spread also has been demonstrated.
The clinical manifestations of HSV disease can be categorized as congenital or neonatal infection, primary infection, or recurrent disease. Currently, neonatal ocular HSV can include conjunctivitis, epithelial or stromal keratitis, cataract, iris atrophy and synechiae, chorioretinitis, optic neuritis, and iridocyclitis.6 The visual prognosis is best for disease that is restricted to the anterior segment, because immune-mediated destruction of these structures is limited in neonates.
In primary disease, the oral mucosa is much more commonly involved than the eye, and the disease is often subclinical or mild. Periocular disease is characterized by a vesicular or ulcerative blepharitis, and ocular disease by a unilateral, acute follicular conjunctivitis that can become pseudomembranous or demonstrate dendrites. Keratitis, which is seen in one third to one half of cases, tends to lag conjunctivitis and lid disease by 1 to 2 weeks. Corneal signs are usually confined to the epithelium and are diffuse and variable in morphology. Infrequently, stromal keratitis or iridocyclitis can occur in primary disease.
Recurrent disease most often affects the cornea, and can affect any or all layers. The characteristic epithelial lesions are dendrites: thin, meandering, arborizing epithelial ulcerations, sometimes with terminal bulbs at the ends of fine branches. Stromal disease may be immune or infectious, or invoke combined mechanisms. The commonly described patterns of stromal herpetic disease include disciform edema, necrotizing stromal keratitis, and immune ring formation. In disciform edema, the dominant feature is disk-shaped stromal edema, without neovascularization or necrosis, that can be focal in milder disease but extensive and diffuse in more severe disease. Folds in Descemet's membrane may accompany corneal thickening, and keratic precipitates accumulate under the area of edema (Fig. 1). Necrotizing stromal keratitis is characterized by inflammatory necrosis and infiltration of the cornea with polymorphonuclear leukocytes, macrophages, lymphocytes, and plasma cells.7 This cellular infiltration produces stromal edema and necrosis. The necrotizing focus can be located at any level of the corneal stroma, with or without epithelial ulcer ation. Ring infiltrates often develop. In some cases, diffuse patches of infiltration appear beneath an intact epithelium, followed by local neovascularization; this is the so-called interstitial keratitis pattern. Lipid deposition, stromal melting, descemetocele formation, and perforation are all potential complications of advanced necrotizing stromal keratitis. If the peripheral cornea is involved, inflammation and necrosis can spread to the sclera, producing a sclerokeratitis.
|
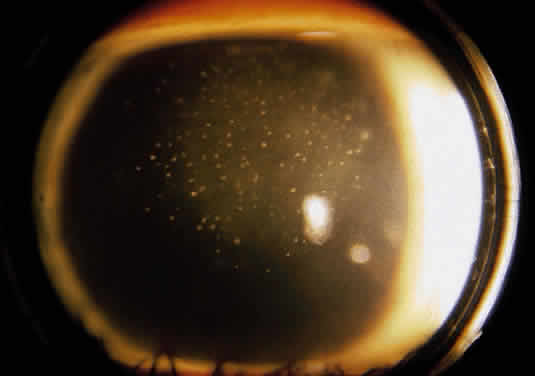
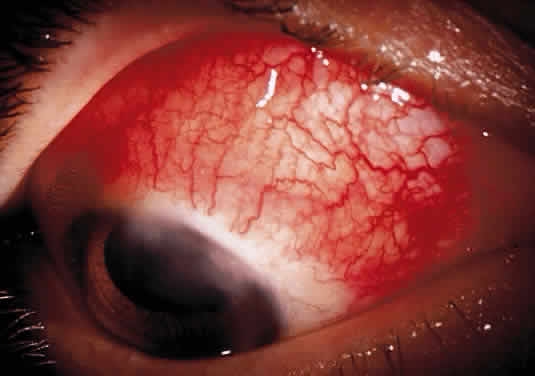
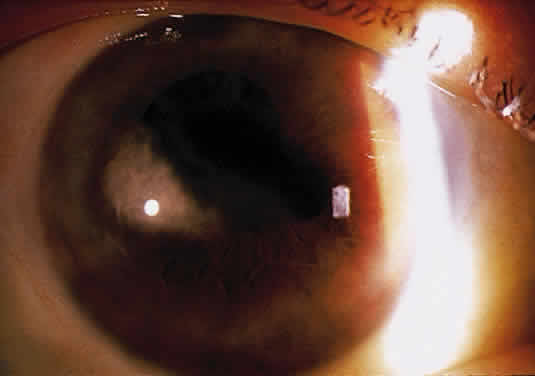
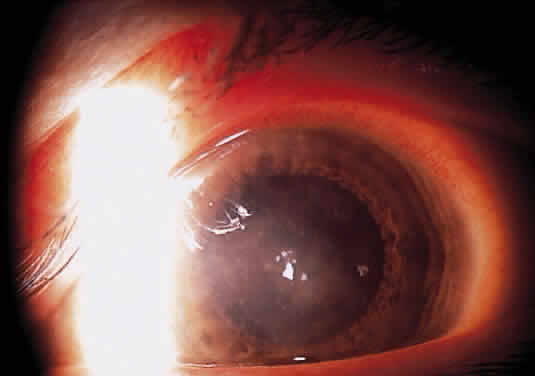
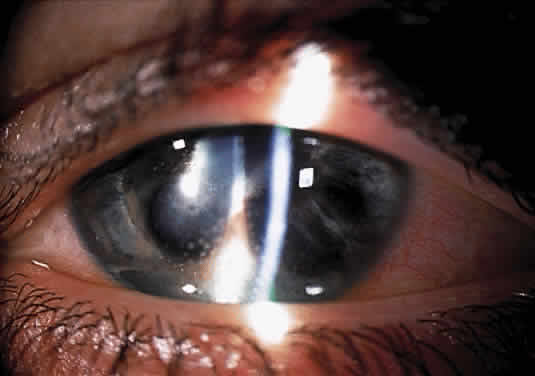
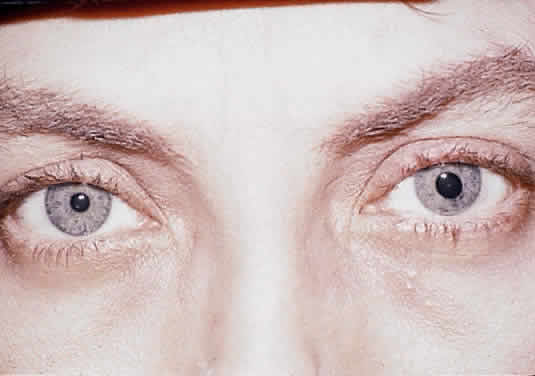
Herpetic keratouveitis can occur in association with any form of herpetic keratitis. Anterior chamber inflammation that accompanies epithelial disease is thought to be caused by reflex irritation and is characteristically both mild and transient. However, the iridocyclitis that invariably accompanies necrotizing disease tends to be much more severe, and is not necessarily correlated with the apparent severity of the keratitis. Indeed, although uveitis dominates the clinical picture in some cases of ocular HSV, subtle corneal findings are limited to faint cellular infiltration of the stroma. Uveitis that accompanies necrotizing disease is typically granulomatous and often recurrent. In more severe cases, perilimbal injection is marked, and the cornea can become thickened and edematous. Dense, fibrinous flare with heavy, anterior chamber cell and medium-sized white keratic precipitates may be distributed widely over the endothelium (Fig. 2). A hypopyon and synechiae can form, and elevated intraocular pressure that exacerbates this often painful uveitis may develop. Marked dilation of iris blood vessels and spontaneous hyphemas sometimes occur. Episodes of inflammation are frequently marked by progressive iris atrophy and sphincter damage, leading to corectopia and anisocoria (Figs. 3 and 4).
|
|
|
Disciform keratitis is usually accompanied by a mild to moderate uveitis. The typical clinical picture is one of regional stromal edema, which directly overlies an area of endothelial inflammatory precipitates, and mild to moderate anterior chamber reaction. A type of HSV-related corneal edema has been observed in which a line of endothelial dysfunction that is reminiscent of corneal allograft rejection develops and progresses from the periphery centrally, accompanied by anterior chamber inflammation and overlying corneal edema.8–10 This endotheliitis may be accompanied by trabeculitis, which leads to elevated intraocular pressure.
PATHOPHYSIOLOGY
Uveitis associated with endotheliitis, disciform keratitis, and necrotizing stromal keratitis can be attributed to both immunologic reaction and live virus invasion. HSV viral particles have been recovered from the aqueous of patients with HSV iridocyclitis.11,12 Nevertheless, in a rabbit model of HSV uveitis, Oh demonstrated that live but not inactivated HSV introduced to HSV antigen-negative animals stimulated a gradual, delayed iridocyclitis, whereas either live or inactivated virus introduced to eyes that had recovered from primary disease rapidly stimulated an inflammatory response.13 These observations suggest that live virus is implicated in the iridocyclitis of primary disease, but that recurrent disease may be mediated by immunologic mechanisms not necessarily dependent upon the presence of live virus. Sundmacher and Neumann-Haefelin have suggested that clinical features such as elevated intraocular pressure, acute focal iritis, and endotheliitis are particularly suggestive of the presence of live HSV in the anterior chamber.12
Live virus also has been isolated from the aqueous of patients with herpetic endotheliitis and stromal edema.8,9 It thus appears that in some cases of disciform keratitis, live virus might infect endothelial cells that demonstrate swelling and pleomorphism. It has been postulated that endothelial damage might result from direct viral cytolysis, as well as from subsequent antigen-antibody-complement (AAC) mediated immunologic attack.12 Most experimental evidence, however, seems to support the theory that endothelial dysfunction and subsequent corneal swelling of disciform keratouveitis is caused by a delayed-type hypersensitivity and immunologic attack directed against endothelial cells that express herpes antigens.12,14,15
TREATMENT
Because the keratouveitis of epithelial HSV is mild and considered reactive, therapy is restricted to treatment of the epithelial disease with a topical antiviral agent, and treatment of the iridocyclitis with a topical cycloplegic agent. Treatment with topical corticosteroids is discouraged because the suppression of the inflammatory response induced by these agents may lead to reduced clearance of antigen, which allows deeper spread of infection and extends the period of exposure to this stimulus, leading to destructive inflammation.16 No evidence suggests that systemic antiviral agents provide added benefit.17
Disciform and necrotizing stromal disease share the potential for irreversible corneal opacification, in the former because of irrevocable endothelial decompensation, and in the latter because of stromal scarring. Associated uveitis also introduces the risk of synechiae formation and glaucoma. All of these conditions must be accounted for in the treatment regimen.
Mild to moderately severe disciform keratitis that is unaccompanied by signs of live virus, such as elevated intraocular pressure or focal iris necrosis, can be treated without the use of topical antiviral agents; however, because it is difficult to prove the absence of live virus, some clinicians prefer to routinely apply a topical antiviral. If concurrent iridocyclitis of any significance is present, the results of a prospective, randomized clinical trial have suggested that oral acyclovir administered for several weeks, along with a topical antiviral agent and topical corticosteroid, may enhance resolution of inflammation.18 These cases tend to be exquisitely responsive to topical corticosteroid administration. Initially, corticosteroids may be applied as frequently as every 2 hours, and topical antiviral agents 4 times a day. Both are usually tapered for several weeks, as signs of inflammation resolve. Cycloplegic agents also should be used to reduce discomfort caused by ciliary body spasm, and to prevent the formation of synechiae. Elevated intraocular pressure should be treated with appropriate agents.
Linear endotheliitis with iridocyclitis is treated in a similar fashion, with topical corticosteroids, topical antiviral agents, and cycloplegic agents. In some cases, oral antiviral medication may prove beneficial in controlling inflammation and preventing a recurrence of disease.10 Associated elevated intraocular pressure should be treated with appropriate medication.
Severe disciform disease and necrotizing stromal disease with significant iridocyclitis can be resistant to therapy. Because there may be live virus present, topical antiviral agents should be used, and agents with enhanced stromal penetration are favored in this situation. Topical acyclovir has excellent stromal penetration and antiviral action but unfortunately is not universally available; alternatives such as trifluridine are commonly used four times a day and tapered over a number of weeks. Inflammatory destruction can be significant, and a prospective, randomized clinical trial has indicated that frequent application (initially eight times a day) of topical corticosteroid effectively shortens the duration of stromal keratitis and reduces persistence or progression of HSV stromal keratitis.19 Because, as noted above, oral acyclovir administered for several weeks may enhance the resolution of herpetic iridocyclitis, systemic antiviral treatment should be administered along with topical medication. In this situation, elevated intraocular pressure is common and should be treated with topical agents. Cycloplegic agents also should be used. Miotics that may contribute to inflammation and synechiae formation should be avoided. Because attacks of severe herpetic keratouveitis are often prolonged, all medicines must be tapered slowly and cautiously, with careful patient follow up.