TABLE 1. Clinical Data on Patients with Cicatricial Pemphigoid
| No. of patients | 108 |
| Mean age (range) | 70 years (43–88) |
| Ratio of women to men | 1.8:1 |
| No. of patients (%) with skin lesions | 17 (16%) |
| No. of patients (%) with lesions of oral mucosa | 45 (42%) |
CUTANEOUS INVOLVEMENT
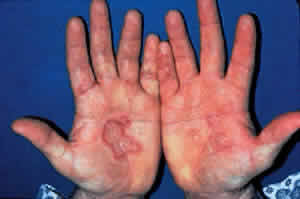
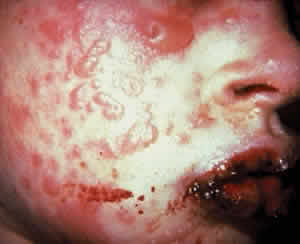
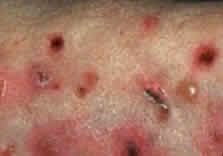
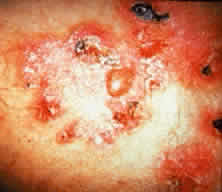
Skin involvement is found less frequently than mucuos membrane involvement (see Table 1) and is reported in 9% to 24% of cases.1–5 The skin lesions of CP may be divided into two types: (1) a recurrent, vesiculobullous, nonscarring eruption that may involve the inguinal area and extremities and occasionally becomes generalized (Fig. 1); and (2) localized, erythematous plaques with overlying vesicles and bullae that appear on the scalp and face near the affected mucous membranes and heal, leaving smooth, atrophic scars (Fig. 2).6
|
|
MUCOUS MEMBRANE INVOLVEMENT
Cicatricial pemphigoid may involve the following mucous membranes: conjunctiva, nose, mouth, pharynx, larynx, esophagus, anus, vagina, and urethra. In a dermatologic study, oral lesions were found in 91% and conjunctival lesions in 66% of patients with CP.5 In ophthalmic studies, 15% to 50% of patients showed involvement of the oral mucosa, whereas 100% had ocular involvement.1–4
CP is associated with two types of oral lesions.6 The first is a desquamative gingivitis that may be patchy or diffuse, heals slowly, and may persist for years. The second type is characterized by vesicles and bullae of the oral mucosa that develop rapidly, remain intact for a few days, and then rupture. Mucous membrane erosions may heal with scarring and even strictures. Stenosis of the nasopharynx or larynx may cause obstructive sleep apnea; esophageal strictures may result in asphyxiation and death when food is swallowed.7
OCULAR INVOLVEMENT
Ocular CP (OCP) is a bilateral disease. The initial symptoms are similar to those of any chronic conjunctivitis and include irritation, burning, and tearing.8 A mucopurulent discharge suggests a complicating bacterial blepharoconjunctivitis. Breakdown of the corneal epithelium leads to foreign-body sensation, photophobia, and reduced visual acuity.
Fibrosis beneath the conjunctival epithelium is the hallmark of CP.9,10 Symblepharons, fibrotic bands that pass between the palpebral and bulbar conjunctiva, involve the inferior fornix first and are best demonstrated early in the disease by drawing the lower eyelid down and having the patient look up (Fig. 3). CP may be associated with a dry eye. Fibrosis beneath the conjunctival epithelium may cause occlusion of the ducts of the lacrimal and accessory lacrimal glands, leading to decreased aqueous tear secretion. The reduced numbers of mucus-producing goblet cells may contribute to an unstable tear film.11 Conjunctival scarring causes lagophthalmos with abnormal blinking and exposure and entropion with trichiasis and distichiasis. All these factors may cause breakdown of the ocular-surface epithelium.
|
Conjunctival or corneal bullae have been described10 but are rarely observed, perhaps because they rupture readily because of the blinking action of the eyelids. Breakdown of the corneal epithelium most commonly results from entropion with trichiasis; lagophthalmos with abnormal blinking and exposure; and a diminished, unstable tear film. These erosions may become complicated by secondary bacterial infiltrates and ulcers. Predisposing factors for the development of microbial keratitis include topical corticosteroids, bandage contact lenses, trichiasis, corneal surgery, lagophthalmos, and meibomianitis.12 Corneal neovascularization may develop either as a pannus or pseudopterygium.
Smears of conjunctival scrapings from patients with CP reveal neutrophils, keratinized squamous cells, and eosinophils.13 In a study of the bacterial flora of patients with CP, potential pathogens—mainly mannitol-positive staphylococci—were recovered from the eyelids and conjunctiva of 81% of patients with this disease.2 Glaucoma may be more prevalent in patients with CP.14
COURSE OF OCULAR DISEASE
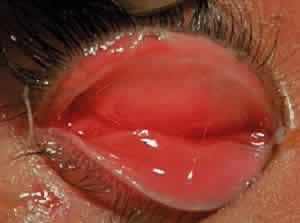
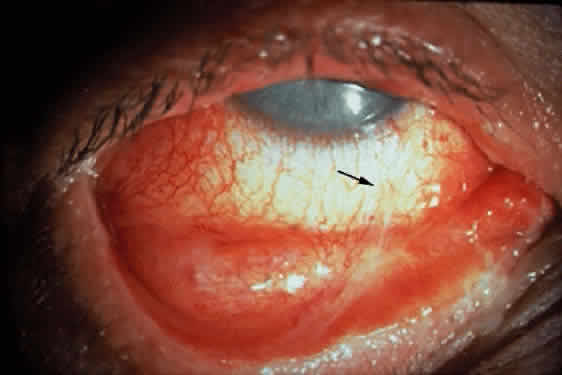
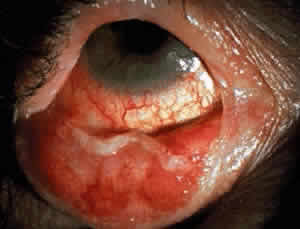
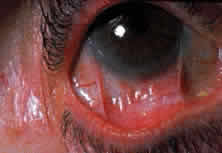
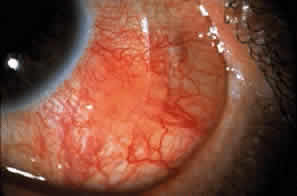
Cicatricial pemphigoid generally runs a chronic course, characterized by progressive shrinkage of the conjunctiva. When the end stage of this disease is reached, the eye lacks tears and has obliterated conjunctival fornices, ankyloblepharon, and a keratinized ocular surface epithelium. Episodes of acute disease activity may interrupt this chronic progressive course and result in rapid shrinkage of the conjunctiva.15 Acute disease activity may be precipitated by surgical procedures, including conjunctival biopsy, lysis of symblepharon, oculoplastic procedures on the eyelids, and cataract extraction. The acute manifestations consist of localized, ulcerated conjunctival mounds (Fig. 4) or diffuse, severe conjunctival hyperemia and edema (Fig. 5). Before concluding that acute inflammatory activity is caused by the disease process, it is necessary to eliminate other confounding factors such as trichiasis, exposure, or bacterial blepharoconjunctivitis.
|
|
When evaluating a patient with CP, the extent of conjunctival shrinkage that has taken place should5 be determined. The most useful positions for examination and photography include upward gaze with the lower eyelid retracted and downward gaze with the upper eyelid retracted. The staging system of Mondino and Brown16 is based on the percentage of conjunctival shrinkage (Fig. 6).2 The staging system of Tauber and coworkers17 describes conjunctival destruction, as follows:
|
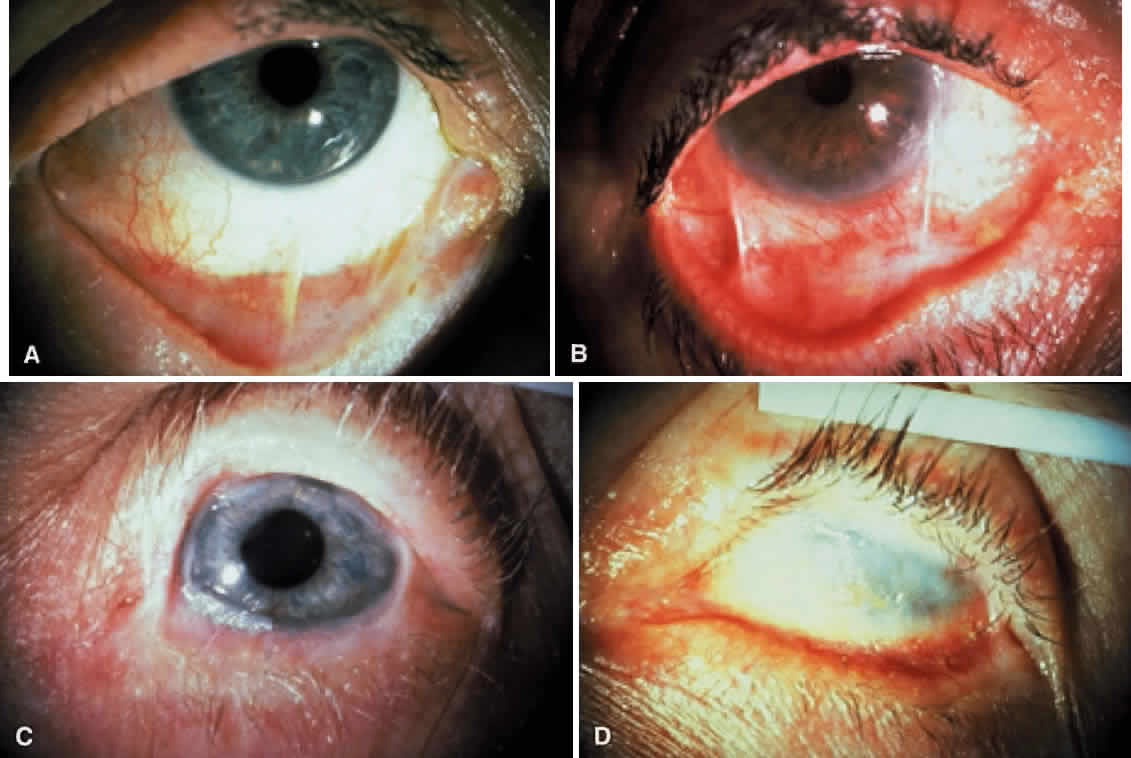
Stage I—chronic conjunctivitis and subepithelial fibrosis
Stage II—early fornix foreshortening
Stage III—symblephara
Stage IV—ankyloblepharon and a frozen globe.
A patient with CP is more likely to have a dry eye, corneal vessels, trichiasis, and positive cultures of the lids and conjunctiva for potential pathogens as the disease advances in stage (Table 2).1
TABLE 2. Correlation of Clinical Data With Stage in Patients With Cicatricial
Pemphigoid
| Stage | No. of Eyes | No. With Dry Eye (%)* | No. With Corneal Vessels (%) | No. With Trichiasis (%) | No. of Positive Cultures of Lid and Conjunctiva for Pathogens† | No. Untreated Showing Progression of Conjunctival Shrinkage‡ (Mean Follow-up 2 Years) |
| I (25% or less shrinkage of conjunctival fornices) | 65 | 19/65 (29%) | 20/65 (31%) | 38/65 (58%) | 22/54 (41%) | 13/32 (41%) |
| II (25% to 50% conjunctival shrinkage) | 62 | 20/62 (32%) | 49/62 (79%) | 52/62 (84%) | 26/55 (47%) | 16/26 (62%) |
| III (approximately 75% conjunctival shrinkage) | 28 | 17/28 (61%) | 28/28 (100%) | 24/28 (86%) | 17/25 (68%) | 8/11 (73%) |
| IV (end stage) | 7 | 7/7 (100%) | 7/7 (100%) | 7/7 (100%) | 5/5 (100%) | - |
| All stages | 162 | 63/162 (39%) | 104/162 (64%) | 121/162 (75%) | 70/139 (50%) | 37/69 (54%) |
*Less than 5 mm of wetting on Schirmer filter strips after 5 minutes without topical anesthesia.
†Mannitol-positive or coagulase-positive Staphylococcus was the most prevalent potential pathogen recovered.
‡Progression defined as any increase in conjunctival shrinkage, including loss of fornix, new symblepharon formation, or extension of symblepharon.
In addition to staging the extent of conjunctival shrinkage that has taken place, it is important to assess the degree of conjunctival inflammatory activity. Stage and inflammatory activity are important parameters to assess but are not necessarily correlated. For example, an eye at stage IV may show no conjunctival inflammatory activity, whereas an eye at stage II may show severe conjunctival inflammatory activity.
Disease progression should be determined by clinical examination and comparison with previous photographs of the external eye.8,16 Progression is defined as increased conjunctival shrinkage—which may involve loss of fornix—new symblepharon formation, or enlargement of an existing symblepharon.
One study suggested that CP may be asymmetric in severity (i.e., both eyes may not be at the same stage) and progression (i.e., both eyes may not necessarily progress or fail to progress).16 Although most eyes progressed, the untreated disease had a variable course because there were eyes in all stages that did not progress. Progression was more likely to occur over a given period in the later stages (i.e., an eye at stage III was more likely to progress than an eye at stage I; see Table 2).
DRUG-INDUCED OCULAR CICATRICIAL PEMPHIGOID
Topical drugs associated with conjunctival scarring include idoxuridine,18 echothiophate iodide,19 pilocarpine,20 demecarium,20 epinephrine,21 and timolol.22 A study of drug-induced conjunctival shrinkage concluded that the spectrum of disease ranges from a self-limited toxic form to a progressive, immunologic form indistinguishable from CP.23 Patients in four studies did not have extraocular involvement of mucous membranes or skin.18–20,23 Patients with drug-induced conjunctival shrinkage may show immunoglobulins bound to conjunctival basement membrane.18,19,24 In a histopathologic study of drug-induced CP affecting the conjunctiva, light and electron microscopy of the conjunctiva revealed findings identical to those previously reported for idiopathic CP.22
There are five possible explanations offered for the relation between drugs and conjunctival scarring:
- The drug may cause conjunctival scarring that mimics CP but is not CP because
the scarring is not progressive.
- The taking of a drug and the development of CP may be coincidental.
- The drug may promote or accelerate the development of CP that may have
developed later anyway.
- The drug may be a cause of CP.
- CP may cause scarring that obstructs collecting vessels of the outflow
pathway and results in glaucoma requiring drug therapy.
A practical approach to the problem of drug-induced conjunctival scarring is to discontinue the topical agents, if this can be done, and follow the patient for progressive conjunctival shrinkage.25 If the drugs are absolutely essential to control glaucoma, they should be continued. If progressive conjunctival shrinkage occurs, the patient probably has CP and immunosuppressant therapy may be warranted. The determination of whether these drugs simply cause scarring that mimics CP or whether they cause or promote CP in a particular patient can be made only by long-term follow-up after drug discontinuation for the development of chronic, progressive conjunctival shrinkage or the development of extraocular lesions.
HISTOPATHOLOGIC FINDINGS
Cicatricial pemphigoid is characterized by subepithelial blisters or bullae. Electron-microscopic studies show that the separation at the margin of a blister is located within the lamina lucida between the plasma membrane of the basal cells and the electron-dense lamina densa.5
The conjunctival epithelium in CP shows squamous metaplasia, with parakeratosis and keratinization (Fig. 7).26 Mucus-producing goblet cells are scarce or lacking.11,27 Because the mitotic rate of conjunctival epithelium is higher in patients with CP, it has been suggested that CP is associated with hyperproliferation of the conjunctival epithelium, with a failure of conjunctival differentiation because of reduced goblet cells.28
|
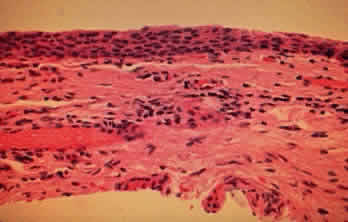
In the early stages of conjunctival disease, granulation tissue is found beneath the conjunctival epithelium, with an infiltration predominantly of lymphocytes and plasma cells, with occasional eosinophils and relatively few neutrophils.13,26 Later, pronounced fibrosis takes place in the conjunctival stroma and is responsible for the conjunctival shrinkage that characterizes the disease.9,10 Hyperproliferation of conjunctival fibroblasts from patients with CP has been demonstrated in tissue culture.29 One study described perivascular inflammatory cell infiltration in 20% of specimens and substantial mast cell participation and degranulation.4 In addition to the chronic inflammatory cells typical of CP, conjunctival biopsy specimens from patients with acute manifestations of CP show numerous neutrophils within and beneath the conjunctival epithelium (Fig. 8).15
|
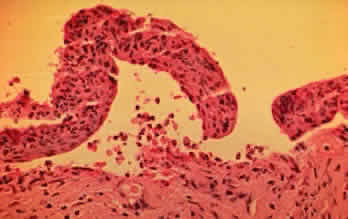
An electron-microscopic study of conjunctival epithelium from patients with CP showed increased desmosomes and prominent tonofilaments and tonofibrils throughout the epithelial cytoplasm. The basal lamina showed areas of discontinuity, duplication, and focal thickening. Collagen fibrils were highly disorganized, and the vascular space was reduced.30 A scanning electron-microscopic study of conjunctival surfaces demonstrated a homogeneous granular sheet of amorphous, mucin-like material covering extensive areas of the conjunctiva in patients with CP that was absent from normal subjects.31
IMMUNOPATHOLOGY
CP is characterized by the binding of immunoglobulins (most commonly IgG) and components of both the classic and alternative complement pathways to the basement membrane zone of skin and oral mucosa.32,33 Circulating antibodies to the basement membrane zone can be demonstrated occasionally, and decreased serum levels of interleukin-6 and increased serum levels of tumor necrosis factor-alpha may be present.33,34 OCP and bullous pemphigoid antigens are distinct but have a common localization within the lamina lucida of the dermal-epidermal junction.35 Patients with systemic features of CP may also have antigens in the lamina densa.36 The bullous pemphigoid antigen is present on the blister roof in close association with basal cells; CP antigen is present on the blister floor.
Conjunctival biopsies taken from patients with CP demonstrate immunoglobulins and complement bound to the basement membrane (Fig. 9) 24,37–43 One targeted basement membrane component may be laminin.44 The destruction of laminin, which normally binds basal epithelium to its basement membrane, leads to blister formation.45 An animal model of subepithelial blistering diseases has been developed using antilaminin antibodies in neonatal mice.46 Further damage causes fragmentation of the basement membrane and subsequent repair with aberrant forms of basement membrane collagen.47 Immunoglobulin deposition on the conjunctival basement membrane also may be found in other diseases.24,38,43,48
|
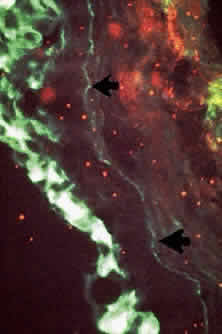
The percentage of patients who demonstrate circulating antibodies to conjunctival basement membrane varies from 0% to 50%.4,37–39,49 In one study, the detection of these antibodies correlated with the degree of clinical activity.49
In CP, antibodies may be directed against specific T-cell antigens expressed in the conjunctival epithelium and its basement membrane (Fig. 10).38,39,43,50 In addition, circulating antibodies that bind to the conjunctival and corneal epithelium have been demonstrated.38,39 Studies showing the presence of transforming growth factor-beta 1 and 3, IL-2, and b-fibroblast growth factor suggest that these cytokines play a role in fibrosis.51,52
|
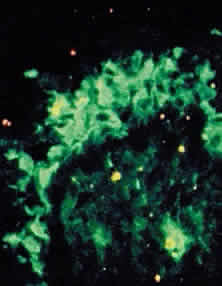
The conjunctiva of patients with CP showed increased numbers of T lymphocytes within the epithelium and substantia propria.53 More helper than suppressor T cells were found. Macrophages were the next most common cells found in the substantia propria, followed by B cells and plasma cells.52 About half of patients with CP have elevated serum IgA levels.38,39 Patients with CP have reduced numbers of circulating T lymphocytes, as determined by E rosette formation.54 The presence of costimulatory molecules may upregulate T-cell activity.55 In one study, CP was associated with HLA-B12, suggesting an immunogenetic susceptibility to the development of this disease.56 Other studies have shown CP to be associated with HLA-DP, HLA-DR4, HLA-DR5, HLA-DQw3, HLA-A2, HLA-B8, HLA-B35, and HLA-B49,57,58 or HLA-DQB1.59,60
DIFFERENTIAL DIAGNOSIS
The clinical diagnosis of CP depends on the documentation of progressive conjunctival shrinkage. The presence of skin and extraocular mucosal lesions suggests the diagnosis. The finding of immunoglobulins and complement bound to the conjunctival basement membrane also support CP as the cause of cicatrization. Immunofluorescence studies on cultured skin keratinocytes may become a corroborative test for circulating antibodies associated with CP.61 Other causes of conjunctival shrinkage and symblepharon must be excluded.
Conjunctival scarring may result from irradiation and severe chemical burns, especially alkali. Symblepharons have also been reported with Sjögrens syndrome,62 atopic keratoconjunctivitis, and sarcoidosis63 but not to the extent and not with the relentless progression of CP. Conjunctival scarring may develop in association with scleral buckles and conjunctival carcinoma but these conditions are usually unilateral, unlike CP. Progressive systemic sclerosis (scleroderma) may be associated with a dry eye and progressive conjunctival shrinkage.64 Trachoma causes conjunctival scarring but this usually begins and predominates in the superior fornix and the upper palpebral conjunctiva.
A membranous conjunctivitis that results in conjunctival scarring may be caused by adenovirus types 8 and 19, a primary infection of herpes simplex virus, diphtheria, or β-hemolytic streptococcus.41,65 The acute self-limited nature of these infections contrasts with the chronic, progressive conjunctival shrinkage found with CP.
Conjunctival shrinkage may be associated with systemic practolol66 or penicillamine67 and topical epinephrine, echothiophate iodide, idoxuridine, pilocarpine, timolol, and dipivefrin.18–23 Whether drug-induced conjunctival shrinkage is self-limited or is actually CP is discussed in the section on “Drug-Induced Ocular Cicatricial Pemphigoid.”
Other bullous diseases usually do not cause much diagnostic confusion with CP (Table 3). Bullous pemphigoid and pemphigus do not usually cause conjunctival scarring.43,68 An acute episode of EM major may cause conjunctival shrinkage but the shrinkage is not chronically progressive, as it is with CP.
TABLE 3. Differential Diagnosis of Bullous Diseases
| Cicatricial Pemphigoid | Pemphigus Vulgaris | Bullous Pemphigoid | Erythema Multiforme | Dermatitis Herpetiformis | |
| Course | Chronic | Chronic | Chronic | Acute, self-limited | Chronic |
| Age at onset | Older than 60 | 40–60 | Older than 60 | 0–30, but may occur at any age | 20–50 |
| Sex preference | Women | None | None | Men | Men |
| Cutaneous involvement | + | + | + | + | + |
| Mucous membrane involvement | + | + | Occasional | + | Rare |
| Ocular involvement | + | + | Occasional | + | Rare |
| Pathology | Subepithelial bulla | Acantholysis, intraepithelial bulla | Subepithelial bulla | Subepithelial bulla | Subepidermal vesicle and bulla |
| Immunofluorescence | + Basement membrane | + Intracellular | + Basement membrane | + Blood vessels | + Basement membrane |
+ , present
TREATMENT
Artificial tears should be used to treat the dry eye associated with CP. Artificial tears without preservatives may be necessary if the preservatives in commercial tear preparations irritate the eyes or if allergies develop to them. Punctal occlusion can be performed if the puncta have not already been occluded by scarring. CP may be complicated by secondary bacterial blepharoconjunctivitis. As mentioned, potential pathogens—especially mannitolpositive staphylococci—were recovered from the eyelids or conjunctiva of 81% of patients with CP.2 Cultures of the eyelids and conjunctiva should be performed at initial examination and at regular intervals. Topical antibiotics should be administered, based on specific antibiotic-sensitivity testing. The staphylococcal blepharitis that frequently accompanies this condition may be treated with eyelid scrubs followed by an antibiotic ointment such as erythromycin or bacitracin. Oral tetracycline or doxycycline may be useful in the treatment of meibomian dysfunction.
In the presence of sufficiently deep fornices, therapeutic soft contact lenses may be used in selected patients to protect the corneal epithelium from trichiasis and drying. Some patients with CP develop large, recurrent corneal epithelial defects that are painful and require almost continuous use of a pressure patch or tarsorrhaphy. In these cases, therapeutic soft contact lenses may keep the corneal epithelium intact and enhance patient comfort. Artificial tears may have to be used frequently to prevent the lens from drying out. Patients who have dry eyes and wear contact lenses are at increased risk of infection and must be followed-up at regular intervals.
Cryotherapy or electrocautery using a hyfrecator may be performed to eliminate trichiasis. Entropion with trichiasis may be corrected early in the disease by oculoplastic surgical techniques. In the advanced stages of the disease, the benefits of oculoplastic procedures, including mucous membrane grafts, may be nullified by the disease. Surgery on the conjunctiva may entail a risk of triggering acute inflammatory activity that may lead to rapid and alarming shrinkage of the conjunctiva.15 Use of intraoperative mitomycin C may prevent this complication.69
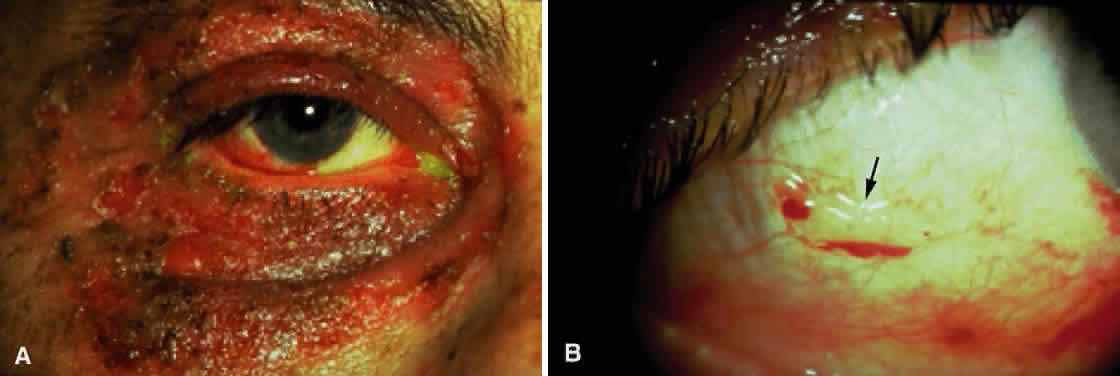
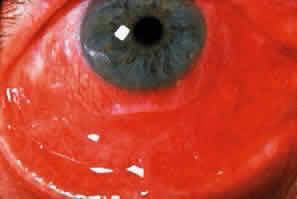
If patients with CP show conjunctival inflammation or evidence of progressive conjunctival shrinkage, cataract surgery or oculoplastic surgery on the lids and conjunctiva should not be attempted until the disease is controlled by systemic immunosuppressant therapy, including corticosteroids. Once disease activity is controlled, mucous membrane grafts may be used to reconstruct or expand the fornices. Favorable results were reported for patients on systemic immunosuppression during cataract surgery.70 Conversely, CP patients who are stable and do not show conjunctival inflammation may not require systemic immunosuppression before surgery but may require postoperative systemic corticosteroids if acute inflammatory activity develops. Systemic corticosteroids are useful in the treatment of the acute manifestations of CP (Figs. 11 and 12).15
|
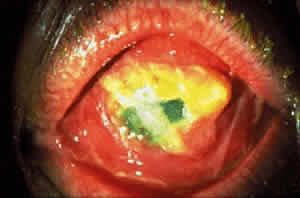
|
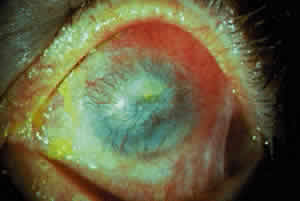
Foster and associates37 and Mondino and Brown16 found that long-term systemic immunosuppressant therapy suppressed conjunctival inflammatory activity and inhibited progression of conjunctival shrinkage. In another report, the results of progression in patients treated with various systemic immunosuppressant agents were compared with those in a control group, with a mean follow-up of about 2 years for all groups (Table 4).1 An eye at stage I or II was less likely to progress and more likely to respond to immunosuppressant therapy than an eye at stage III. Not all patients responded to immunosuppressant therapy, however, and some developed complications that required tapering or even discontinuation. Therefore, older patients should not be subjected to the risk of immunosuppressant therapy unless their eyes demonstrate ongoing progression of conjunctival shrinkage. In addition to being followed-up by an ophthalmologist, patients taking immunosuppressant agents should be monitored by an internist, oncologist, or rheumatologist who is familiar with the toxicity and side-effects of these potent drugs. If immunosuppressant therapy is discontinued, the patient should be monitored for recurrence.71
TABLE 4. Number of Eyes Showing Progression of Conjunctival Shrinkage* in
Control and Treatment Groups (Mean Follow-up, 2 Years)
| Group | No. of Patients | No. of Eyes | Stages | ||
| I | II | III | |||
| Azathioprine | 10 | 20 | 1/3 >(3%) | 5/9 (56%) | 4/8 (50%) |
| Cyclophosphamide | 13 | 26 | 1/4 (25%) | 1/10 (10%) | 9/12 (75%) |
| Prednisone | 11 | 22 | 0/9 (0%) | 1/7 (14%) | 3/6 (50%) |
| Cyclophosphamide and Prednisone | 17† | >33 | 1/6 (17%) | 4/19 (21%) | 2/8 (25%) |
| Treatment Groups Combined | 51† | 101 | 3/22 (14%) | 11/45 (24%) | 18/34 (53%) |
| Control | 35† | 69 | 13/32 (41%) | 16/26 (62%) | 8/11 (73%) |
*Progression defined as any increase in conjunctival shrinkage including loss of fornix or new symblepharon formation.
†One eye from one patient was excluded from analysis because it was already at stage 4.
Dapsone has been used to treat CP.72 It should be avoided in patients who have a history of sulfa allergy or who are glucose-6-phosphate dehydrogenase-deficient. Tauber and associates73 believe that dapsone may be the best initial therapy for patients with CP that shows mild to modest inflammatory activity and is not rapidly progressive, whereas cyclophosphamide may be the best initial choice for highly active cases. Cyclophosphamide may also be combined with systemic prednisone in difficult cases.74 Sulphapyride may also be useful for treating CP.75
In the final stages of CP, when the conjunctival fornices are obliterated and the ocular surface epithelium is keratinized, a keratoprosthesis may be inserted to restore some sight to these patients.76