All immune-related cells arise from a common pluripotential stem cell. This stem cell gives rise to committed stem cells for the lymphoid and myeloid series. Under the influence of particular colony-stimulating factors, the lymphoid and myeloid stem cells give rise to various cell types. The lymphoid cell line consists of lymphocytes such as T cells, B cells, and large granular lymphocytes (see Third Population Cells). The myeloid cell line consists of nonlymphoid cells such as erythrocytes, monocytes/macrophages, polymorphonuclear granulocytes (neutrophils, basophils, eosinophils), megakaryocytes/platelets, and mast cells. The bone marrow is the site of maturation of most of these cell types. A major exception is the T cell, which leaves the bone marrow and matures under thymic influence.
Before continuing with a discussion of immune cell types and function, two topics will be reviewed: the major histocompatibility gene complex (MHC) and cell markers.
MAJOR HISTOCOMPATIBILITY GENE COMPLEX
Alloantigens are antigens that differentiate between members of a species (i.e., intraspecies). The histocompatibility antigens are alloantigens that play a role in the rejection of allogeneic tissue grafts. These surface molecules are coded in the human genome in the major histocompatibility gene complex (MHC). This localized group of genes codes for cell surface antigens, which play a major role in immunity, self-recognition, and tissue graft rejection. Human leukocyte antigen (HLA) is the name for the human MHC. The HLA region is located on the 21 region of the short arm of chromosome 6 (Fig. 1). In addition, there are minor histocompatibility antigens coded throughout the genome that play a definite but less well-defined role in the immune response. There are three classes of gene products of the MHC.2
|

Class I Molecules
These antigens are expressed on virtually all nucleated cell surfaces, including B cells, T cells, and platelets, but not on mature red blood cells. They are products of the HLA-A, HLA-B, and HLA-C loci. Each individual has six serologically defined HLA-A, HLA-B, and HLA-C antigens, three from each parent.
Class II Molecules
These antigens are a product of the HLA-D region, which includes the subregions -DR, -DQ, and -DP. The genes of the HLA-D region that code for class II determinants are known as immune response (Ir) genes. Class II molecules, often called Ia (immune-associated) antigens, are expressed on antigen-presenting cells (monocytes/macrophages, Langerhans cells, dendritic cells), B lymphocytes, and activated T lymphocytes. Immune suppressor (Is) genes also exist in the HLA-DQ region.
Class III Molecules
These antigens include the complement components C4, C2, and factor B.
Although HLA antigens do not directly cause disease, certain HLA types are known to predispose individuals to particular autoimmune diseases (e.g., HLA-B27 and ankylosing spondylitis). The mechanism is not clearly defined but involves immunologic alteration.
SURFACE MARKERS
Cell surface molecules are identifiable by specific monoclonal antibodies. The CD system is presently the standard nomenclature (CD stands for cluster designation). Surface markers can be used to differentiate immune cell types, stages of maturation of a given immune cell type, and immune cell activation.
T-Cell Markers
Pan-T cell markers include CD2, CD5, and CD7 (Table 1). CD2 is responsible for the binding of T cells to sheep erythrocytes forming rosettes (B cells cannot do this). This was formerly the major means of identification of T cells.
TABLE 1. Important T-Cell Markers
| CD Group | Other Names | Cell Type/Function |
| CD1 | T6 | Immature cortical thymocytes, Langerhans cells |
| CD2 | T11, sheep erythrocyte receptor | Original T-cell marker (rosette formation with sheep RBC); T-cell adhesion, activation |
| CD3 | TCR-associated molecules | |
| CD4 | T4 | T helper; MHC class II restriction |
| CD5 | Pan-T cell, some B cells | |
| CD7 | Earliest marker of T-cell line; T-cell activation;pan-T cell, including all third population cells;?receptor for Fc portion of IgM | |
| CD8 | T8 | Suppressor/cytotoxic T cells; class I restriction |
| TCR | Recognizes antigen; T-cell activation induced by antigen | |
| CD29 | 4B4, fibronectin receptor | Present on helper/inducer T cells |
| CD45R | Present on suppressor-inducer T cells (i.e., CD4+ cells that induce suppressor/cytotoxic CD8+ cells) |
RBC, red blood cells; TCR, T-cell antigen receptor; MHC, major histocompatibility gene complex
The T-cell antigen receptor (TCR) is the definitive T-cell marker.3 There are two types of TCR, TCR1 and TCR2, the latter being the most common (present on 95% of T cells). TCR2 is composed of two peptide chains, alpha and beta, linked to form a heterodimer. This is closely associated with CD3 molecules (CD3 subunit), forming the TCR2 receptor complex. TCR1 is similar in structure to TCR2 but contains gamma and delta polypeptides.
The TRC2 group can be divided into those cells that are CD4+ (helper T cells) and those that are CD8+ (suppressor/cytotoxic T cells). Helper T cells recognize antigen associated with class II molecules. The functions of CD4+ cells include promotion of T-cell and B-cell differentiation, regulation of suppressor/cytotoxic T-cell proliferation/function, and production of lymphokines (see below). They also play a role in regulation of erythropoiesis. Those TCR2 cells associated with CD8+ are suppressor/cytotoxic T cells. These cells recognize antigen associated with class I molecules and function as cytotoxic cells. Also, they act as suppressors of T- and B-cell function.
B-Cell Markers
The classic B-cell marker is surface immunoglobulin produced by the cell itself. These immunoglobulins act as B-cell receptors for antigen. On circulating B cells, the surface immunoglobulins are predominately IgM and IgD molecules, and, for a given cell, they share the same antigen specificity. CD19, CD20, and CD22 are presently the markers commonly used to identify B cells.3 Other markers found on B cells include MHC class II antigens, which are important in cooperation with T cells, complement receptors CR1 (for C3b) and CR2 (for C3d), CD5 (also expressed on all T cells), and surface receptors for the Fc portion of IgG.
CELLS OF THE IMMUNE SYSTEM
T Lymphocytes
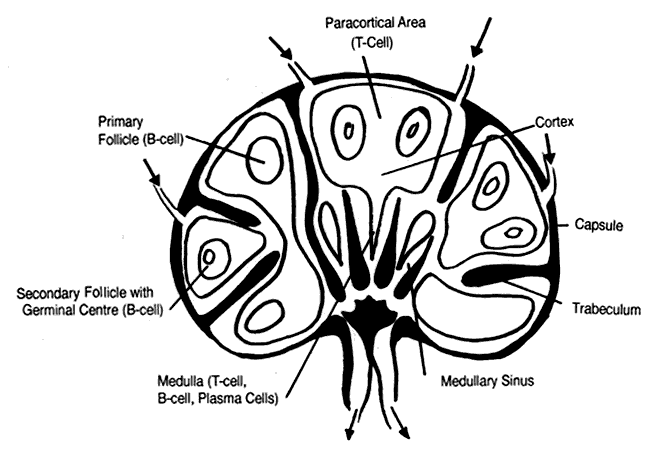
T cells regulate the immune response, are the major players in cell-mediated immunity, and induce antibody production by B cells. T-cell precursors arise from stem cells in the yolk sac, the fetal liver, and, later in prenatal development, the bone marrow. These immature cells migrate to the thymus, where maturation follows. Mature lymphocytes migrate via the circulation to the spleen, lymph nodes, tonsils, and unencapsulated lymphoid tissue. The latter includes mucosa-associated lymphoid tissue (MALT), such as that in the gut (GALT), bronchi (BALT), and conjunctiva (CALT). Lymphocytes make up 20% of the total circulating leukocyte count. Mature T lymphocytes make up 70% to 80% of peripheral blood lymphocytes. In lymph nodes, T cells reside in deep paracortical areas around B-cell germinal centers (Fig. 2) and in periarteriolar areas of white pulp of spleen. The T-cell pool is maintained throughout the years by antigen-driven expansion of long-lived T cells that reside in the peripheral lymph organs. There are two major subsets of T lymphocytes--helper T cells and suppressor/cytotoxic T cells.
|
HELPER T CELLS.
Helper T cells are CD4+ lymphocytes. They are class II restricted (i.e., they respond to antigens presented in conjunction with class II molecules on the surface of antigen-presenting cells [APC], such as macrophages). The CD4 molecule has an adhesive function and binds to the class II (HLA-DR, -DQ, or -DP) molecule on the surface of the APC. This enables TCR to bind to the antigen in a stable fashion. Helper T cells function as regulator/modulator cells. Those CD4+ cells that display the helper function (i.e., positively influence the immune response of T cells and B cells) are CD29+ . These cells induce B cells to produce antibodies. A subset of CD4+ cells that are CD45R+ display the suppressor-inducer function (i.e., induce suppressor/cytotoxic function in CD8+ cells, which results in suppressed antibody production by B cells).4
SUPPRESSOR/CYTOTOXIC T CELLS.
Suppressor/cytotoxic T cells are CD8+ and are class I restricted (i.e., cytotoxic T cells recognize antigens presented in association with class I [HLA-A, -B, or -C] molecules). CD8+ binds to the class I molecule of the antigen-presenting cell. This stabilizes TCR binding to antigen on the surface of the antigen-presenting cell. When this condition is met, lysis of the target cell can occur (see below). These cells play an important role against intracellular parasites and viruses. Suppressor cells may act on either T cells or B cells and perform a down-regulating function. Suppressor/cytotoxic T cells may be activated by helper T cells that are CD45R+ (see suppressor-inducer function above). No surface markers are yet known that distinguish between CD8+ suppressor and cytotoxic cells.
Helper T cells constitute 45% of circulating lymphocytes and suppressor/cytotoxic T cells 25% of circulating lymphocytes, giving a normal helper T/suppressor T ratio of 1.8:1.
B Lymphocytes
B cells are responsible for humoral immunity (i.e., that arm of the immune response mediated by antibodies). B lymphocytes arise from precursor cells in the bone marrow (in birds, in the bursa of Fabricius, hence the name B cell). Mature B cells express surface immunoglobulins, which, as previously discussed, act as receptors for foreign antigen. Each B cell produces only one type of immunoglobulin heavy and light chain variable region; thus, each B cell has specificity for only one antigen.
B cells constitute 10% to 15% of circulating lymphocytes. In lymph nodes, B cells are located in cortical germinal centers and medullary cords, where they are the principal cell type (see Fig. 2). B cells are located in primary and secondary germinal centers of the white pulp of the spleen.
When mature unprimed B cells are stimulated by antigen, they either develop into plasma cells that produce antibody or become memory cells. This is known as the primary response. Memory cells are long lived and respond when re-exposed to the same antigen in the future, enabling an amplified secondary response. Memory cells have higher-affinity antigen receptors and are prone to make IgG earlier than unprimed B cells.5
Plasma cells are restricted in location to tissues and are not found to any significant extent in the circulation. These cells possess an eccentric round nucleus with chromatin arranged in a cartwheel pattern. The cytoplasm is basophilic; large amounts of RNA are present in the rough endoplasmic reticulum, which is actively involved in antibody synthesis. Each plasma cell produces only one class of immunoglobulin, specific for a particular antigen.
Third Population Cells
Third population cells make up 5% to 10% of peripheral blood lymphocytes. These cells typically possess granular morphology and are called large granular lymphocytes. They lack the usual lymphocyte antigen receptors of T and B lymphocytes (i.e., TCR and immunoglobulin, respectively) and thus are also known as non-T, non-B cells, or null cells.3 The definitive marker for this group of cells is CD16. Third population cells do share some common markers with T cells (e.g., CD2, CD7), and to a lesser extent cells of the myelomonocytic series, but are CD4-(30) and often CD8 -(31). Third population cells also express interleukin-2 (IL-2) receptor and thus can be activated by IL-2. Third population cells probably develop in bone marrow, but their sequence of differentiation is not well defined.
Large granular lymphocytes have the ability to kill certain tumor cells and virus-infected cells by NK (natural killer) activity, and target cells primed with IgG by antibody-dependent cell-mediated cytotoxicity (K cells). Also, these cells may release cytokines (e.g., interferon-γ [IFN-γ]), which are important in regulation of the immune response.
Natural Killer (NK) Cells
Natural killer activity describes the non-antibody-mediated, nonphagocytic killing of target cells when there has been no previous contact. Natural killer cells have the ability to recognize the altered surface of infected or neoplastic cells, bind to those cells, and cause lysis of the target cell (i.e., cytotoxicity). The term natural killer describes a function, not an exclusive cell type. Large granular lymphocytes (LGL) are the major NK cells (also note that LGL can function in antibody-dependent cell-mediated cytotoxicity). Other NK cell types include macrophages/monocytes.
Killer (K) Cells
K cells participate in antibody-dependent cell-mediated cytotoxicity. K cells possess surface Fc receptors, allowing binding to the Fc portion of IgG bound to the target cell surface. Binding results in cell lysis.
Monocytes/Macrophages
Monocytes arise from myeloid precursor cells in the bone marrow under the influence of monocyte colony-stimulating factor (M-CSF), granulocyte/macrophage colony-stimulating factor (GM-CSF), and IL-3 (multi-CSF). Mature monocytes circulate in the blood (3% to 8% of peripheral blood leukocytes) and migrate into peripheral tissues, where they are termed macrophages. Usual locations of tissue macrophages include skin, connective tissue, perivascular connective tissue, peritoneum, pleura, synovium, lung, spleen, and lymph nodes. In the liver, they are known as Kupffer cells, in bone as osteoclasts, and in the central nervous system (CNS) as microglia.
Macrophages play an important role in the immune response. Macrophages can function as phagocytes (i.e., can engulf particles and destroy them) by binding via Fc receptors to the Fc portion of antibody bound to the target cell, by binding to complement on the target cell via complement receptors, or by engulfing without opsonization. This is not cytotoxicity (i.e., NK activity) by definition. However, macrophages also do possess NK activity. Macrophages are important in antigen presentation to lymphocytes. Their role also includes immunoregulation by monokine production. The most important example is T-cell activation promoted by interleukin-1 (IL-1) secreted by macrophages. Tumor necrosis factor (TNF) is another important monokine.
Macrophages possess surface class II molecules, and with activation, the expression of class II molecules increases. In addition to Fc receptors and complement receptors (CR1 and CR3), macrophages possess surface receptors for migration inhibitory factor (MIF) and macrophage activation factor (MAF), IFN-γ, and other lymphokines, all of which play a role in macrophage activation. IL4 and transforming growth factor-ß (TGF-ß) antagonize macrophage activation.
Granulocytes
Granulocytes arise from precursors in the bone marrow and include neutrophils, eosinophils, and basophils. Granulocytes are found in the circulation, where they constitute 60% to 70% of total blood leukocytes. They are also present in extravascular tissues.
NEUTROPHILS.
Neutrophils make up 90% of the granulocyte population. Neutrophils spend a relatively short time in the circulation with a half-life of 6 to 7 hours; they survive longer in extravascular tissues, dying after 1 to 4 days. Neutrophils express receptors for C5a and other chemotactic factors.
These chemotactic factors stimulate migration of neutrophils from the blood into extravascular sites via margination and diapedesis. Neutrophils also express Fc receptors for IgG and receptors for C3b; these facilitate binding to microorganisms and subsequent phagocytosis. Engulfed microorganisms are contained in vacuoles called phagosomes, which fuse with lysosomes (see below) to form phagolysosomes, in which the foreign microorganisms are digested. Lymphocyte function-associated antigen type 1 (LFA-1) is found on neutrophils as it is on all leukocytes. LFA-1 mediates intercellular adhesion between leukocytes and other cells (non-antigen specific).
Neutrophils possess primary granules (lysosomes) that contain acid hydrolases, myeloperoxidase, elastase, lysozyme (muramidase), and other enzymes. Also, there are secondary granules that contain lactoferrin, lysozyme, collagenase, and others. Neutrophils are important in type III hypersensitivity reactions, binding to the Fc portion of immune complexes with subsequent degranulation. The extracellular release of enzymes is followed by generation of superoxide radicals that kill miocroorganisms but also lead to injury to surrounding tissue. Persons with abnormal neutrophil function are particularly prone to pyogenic infection.
EOSINOPHILS.
Eosinophils constitute 2% to 5% of blood leukocytes. These granulocytes possess a bilobed nucleus and numerous acidophilic granules. Eosinophils were classically believed to down-regulate the immune response because of anti-inflammatory enzymes such as histaminase (inactivates histamine), arylsulfatase (inactivates slow-reacting substance of anaphylaxis [SRS-A]), and phospholipase D. However, eosinophils are now recognized as playing a more active role in inflammatory and allergic processes.
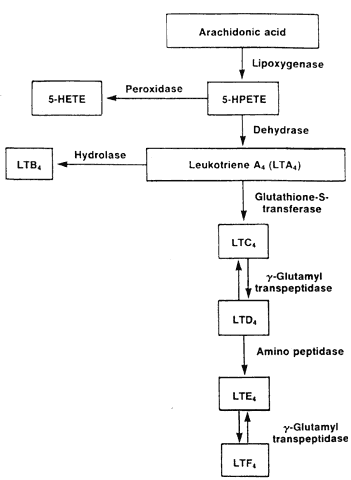
Eosinophils possess receptors for complement fragments and the Fc portion of IgG, IgM, and IgE. Eosinophils are chemotactic to platelet activating factor (PAF) and eosinophilic chemotactic factor of anaphylaxis (ECF-A), both of which are produced by mast cells. Eosinophils are capable of phagocytosis but also possess cytotoxic abilities achieved through degranulation. Eosinophilic granules contain cytotoxic basically charged proteins, including eosinophilic major basic protein (EMBP), eosinophilic cationic protein, and eosinophil peroxidase. Each is toxic to schistosomes and other parasites. EMBP is particularly important in allergic diseases such as asthma and vernal keratoconjunctivitis because of its ability to stimulate mast cell degranulation and damage host tissue.6 Eosinophils generate lipid-derived mediators (e.g., leukotriene C4 and PAF).
BASOPHILS.
Basophils make up less than 0.2% of circulating leukocytes. Basophils are also found in tissue and like mast cells play a role in allergic mechanisms and cutaneous basophil hypersensitivity (Jones-Mote). IgE receptors on the basophil surface bind IgE. Antigen binding stimulates release of inflammatory mediators similar to those of mast cells (see below).
Mast Cells
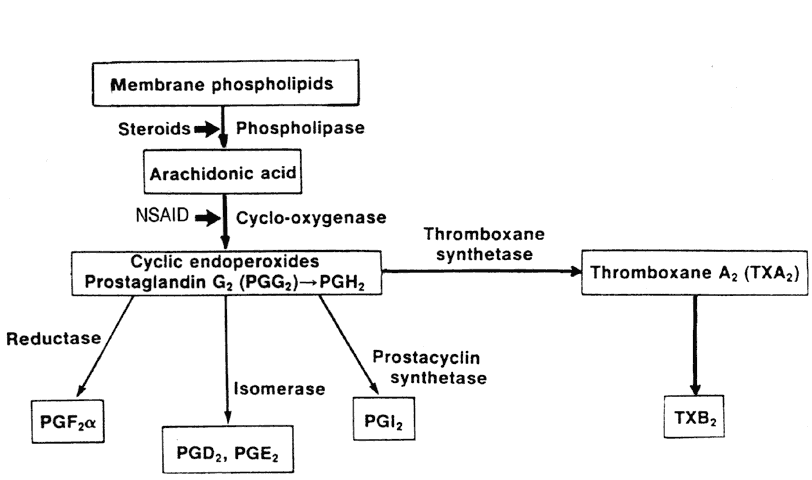
Tissue mast cells play the major role in type I hypersensitivity reactions. The mast cell membrane has as many as 500,000 IgE receptors, 10% of which are occupied in vivo.7 The Fc portion of the IgE molecule binds to these receptors, leaving the Fab portion exposed to bind antigen. With antigen binding, calcium influx ensues and the mast cell immediately releases preformed chemical mediators located within granules (e.g., histamine, ECF-A, neutrophil chemotactic factor). In addition, the arachidonic pathway is stimulated, resulting in de novo synthesis of prostaglandins and leukotrienes. PAF, a potent eosinophilic chemotactic factor, is also produced. Mast cell degranulation is believed to be responsible for the immediate wheal and flare response in skin tests and arachidonic acid metabolites, the late-phase response.
A negative feedback mechanism exists to keep the reaction in check. Released histamine binds to histamine receptors on the surface of mast cells, leading to activation of adenyl cyclase, which converts ATP to cyclic AMP (cAMP). Increased cAMP turns off mast cell degranulation, resulting in a negative feedback control. Beta-adrenergic receptor activation and certain prostaglandins also activate adenyl cyclase and increase cAMP levels. The enzyme phosphodiesterase degrades cAMP; thus, phosphodiesterase inhibitors such as theophylline play a beneficial role in allergic disease by maintaining levels of cAMP. Alpha-adrenergic receptor stimulation decreases cAMP levels. Cyclic GMP (cGMP) also modulates the mast cell response, having the opposite effect of cAMP. Increased cGMP levels stimulate mast cell degranulation mediator release. Cholinergic stimulation increases the concentration of cGMP.
Antigen-Presenting Cells
T cells cannot recognize free antigen. Suppressor/cytotoxic T cells recognize antigen in association with MHC class I molecules on the surface of target cells (i.e., a class I-restricted phenomenon). Helper T cells recognize antigen presented in association with MHC class II molecules on the surface of antigen-presenting cells (class II-restricted) (Fig. 3).
|
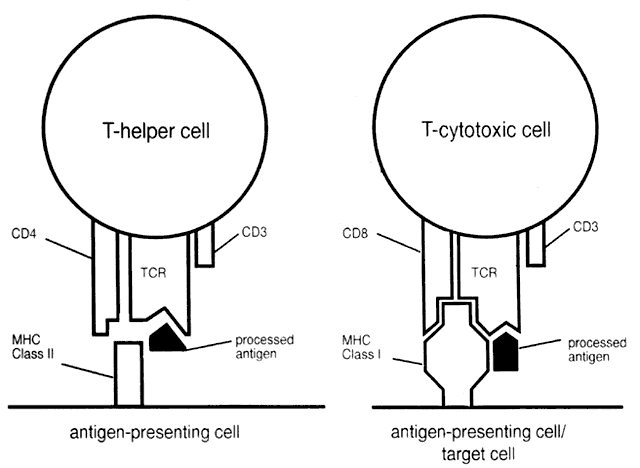
Antigen-presenting cells pick up antigen, which is then processed (i.e., large antigen complexes or cells are degraded into an optimal size of 8 to 24 amino acids to allow fitting into HLA surface molecules) and presented on the surface in conjunction with HLA class II molecules. This enables helper
T cell binding and activation. Phagocytes (monocytes/macrophages), B cells, and dendritic cells (including Langerhans cells) can all present antigen to class II-restricted helper T cells. In addition, cells that do not usually express class II MHC antigen, in particular vascular endothelial cells, can be induced by IFN-γ and TNF to express class II MHC on their surfaces and thus can present antigen to helper T cells. This phenomenon may play a role in autoimmune disease.
Langerhans cells are bone marrow-derived cells normally present in the epidermis, conjunctiva, and corneal limbus. They possess characteristic Birbeck granules in the cytoplasm. These cells travel via the afferent lymphatics to the local lymph nodes, where they settle in the paracortical areas and interdigitate (thus called interdigitating dendritic cells) with T cells, thereby presenting antigen to T cells in the draining nodes. Langerhans cells are rich in class II MHC molecules and thus are particularly involved with presenting antigen to CD4+ cells.
LYMPHOCYTE ACTIVATION
The key to activation of both T and B lymphocytes in vivo is antigen binding. Antigen is processed by antigen-presenting cells that present antigenic peptides (epitopes) in association with class II MHC molecules on their surface (Fig. 4). This is recognized by the helper T-cell receptor (first signal), resulting in helper T-cell activation and proliferation. This process is enhanced by IL-1 (second signal) produced by macrophages. The resulting increase in helper T-cell function induces cytotoxic T cells and other effector cells and B-cell differentiation. In addition, B cells themselves are able (in the absence of T cells) to recognize directly both free antigen and antigen presented on the surface of antigen-presenting cells by binding via surface immunoglobulin. However, T cells are required for full B-cell response. The result is B-cell stimulation, plasma cell differentiation and antibody production, and memory cell formation.
|
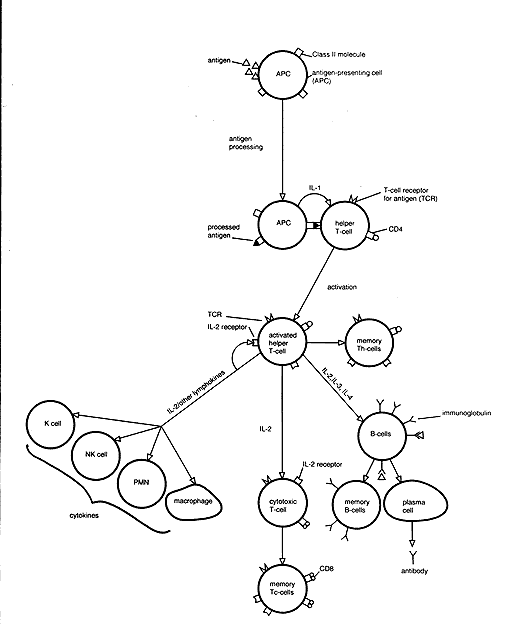
T and B lymphocytes carrying specific antigen receptors exist in the body as a resting lymphoid pool. When stimulated by a specific antigen for the first time, these cells proliferate and differentiate into effector cells (plasma cells from B cells; helper and/or suppressor/cytotoxic cells from T cells) and memory cells (memory B cells, memory T cells). This initial response is called the primary response. Memory cells are long lived and respond when re-exposed to the same antigen in the future. When the memory cells are subsequently rechallenged by the same specific antigen, they are capable of generating a more efficient and magnified response, known as the secondary response (anamnestic response). Again, both effector and memory cells are produced, but in greater numbers. The antibodies produced in a secondary response appear more quickly, consist predominantly of IgG, attain a higher titer, and have a higher affinity for antigen than those produced in the primary response.
Activation causes changes in surface markers. IL-2 receptors and class II (HLA-DR) molecules are expressed with T-cell activation and are major markers for this activation. B-cell activation markers include receptors for IL-2 as well as IL-3, -4, -5, -6, and transferrin, as well as HLA-DR (class II) molecules.
CELL-MEDIATED IMMUNITY
The term cell-mediated immunity (CMI) includes those immune mechanisms in which cellular interactions predominate and antibody, although not totally excluded, plays a lesser role. The key player in cell-mediated immunity is the helper T cell, which, when activated by a specific antigen, modulates the activity of various other immune cells. B cells are stimulated to produce antibody. Cytotoxic T cells, NK cells, K cells, macrophages, and granulocytes are influenced by helper T cells and participate in the cell-mediated responses. Cytokines produced by these cells influence the immune processes. T-suppressor function dampens B-cell activity and the function of other T cells. The major components of CMI include antigen presentation and lymphocyte activation (described previously), cell-mediated cytotoxicity, macrophage involvement, and cytokine production.
Cell-Mediated Cytotoxicity
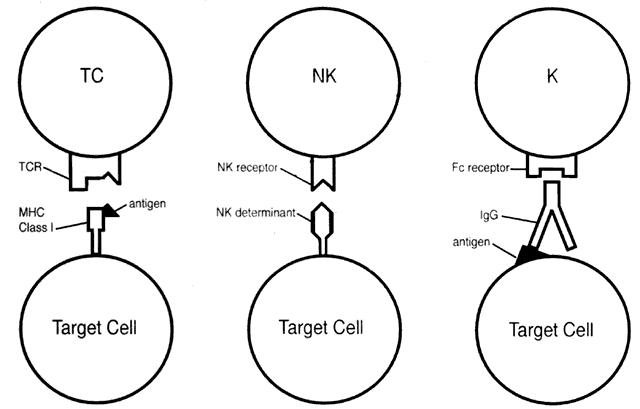
Lymphoid and some myeloid cells can bind to and lyse target cells (not phagocytosis). Cytotoxicity occurs by three main mechanisms (Fig. 5): cytotoxic T-cell activity, antibody-dependent cellmediated cytotoxicity, and natural killer (NK) activity.
|
CYTOTOXIC T-CELL ACTIVITY.
Via their MHCrestricted T-cell receptors, cytotoxic T cells recognize antigen in conjunction with class I MHC molecules present on the surface of target cells. The result is lysis of the target cell. Although the majority of cytotoxic cells are CD8+ , 10% of MHC-restricted cytotoxic T cells are CD4+ and thus class II restricted. An important role of cytotoxic T cells is elimination of virus-infected cells.
ANTIBODY-DEPENDENT CELL-MEDIATED CYTOTOXICITY.
This form of cytotoxicity involves K cells that possess surface Fc receptors. The K cell Fc receptor binds to the exposed Fc portion of antibody (IgG) bound to antigen on the surface of the target cell. The result is cell lysis. Fc receptors are found on macrophages, neutrophils, and large granular lymphocytes (LGL).
NATURAL KILLER (NK) ACTIVITY.
Most NK cells are LGL. These cells are able to recognize nonspecific and MHC-unrestricted determinants on the surface of targets cells and thereby induce cell lysis. Lymphokine-activated killer cells (LAK) are cells activated by culturing in high levels of IL-2; these cells also exhibit NK activity. K and NK activity can be properties of the same cell.
Macrophage Functions in CMI
As mentioned previously, macrophages are involved in initiation of lymphocyte activation by presenting processed antigen and production of IL-1. Macrophages are also effector cells. Activated helper T cells release lymphokines such as macrophage activation factor IFN-γ and migration inhibition factor, resulting in macrophage stimulation. The resulting macrophage effector functions are numerous. Macrophages play a role in inflammation and fever, microbicidal and tumoricidal activity, and tissue damage and repair. These functions are achieved by various mechanisms, including phagocytosis, NK activity, monokines (e.g., IL-1, TNF), enzymes (e.g., acid hydrolases, proteases, lysozyme, elastase, hyaluronidase, collagenase), products of oxidative metabolism (H2O2), complement, and prostaglandins.
Cytokines
Cytokines8 include both lymphokines and monokines, produced by lymphocytes (primarily T cells but also B cells) and macrophages, respectively, as well as mediators secreted by other cells (NK, LGL). These soluble molecules are intercellular messengers and have in common the function of mediating the action and interaction of cells involved in the immune response. Cytokines may work alone or in cooperation with other cytokines. Some of the more important cytokines are discussed below (Table 2).
| Cytokine | Other Names | Source | Function |
| Interleukin-1 (IL-1) | Lymphocyte activating factor, leukocytic pyrogen | Macrophages, LGL, B cells | Macrophage stimulation; lymphocytedactivation; increased NK activity;dchemoattractant for PMNs, lymphocytes; induces fever |
| IL-2 | T cells | T- & B-cell proliferation | |
| IL-3 | Multi-CSF | T cells | Stimulates hematopoietic stem cells of several lines (neutrophils, monocytes, eosinophils, basophils, erythrocytes, megakaryocytes) |
| IL-4 | T cells | B-cell & T-cell proliferation; regulation of IgE synthesis | |
| IL-5 | T cells | B-cell & eosinophil differentiation | |
| IL-6 | T cells, B cells, macrophages | B-cell growth, differentiation; acute-phase proteins | |
| IL-7 | Bone marrow stromal cells | Growth of B-cell & T-cell precursors | |
| IL-8 | Fibroblasts, monocytes, endothelium | Chemotactic for PMNs, T lymphocytes; promotes adhesion of PMNs to vascular endothelium | |
| Tumor necrosis factor (TNF) | Lymphocytes, macrophages | Macrophage, granulocyte & cytotoxic cell activation; enhances MHC expression; tumoricidal activity; circulatory collapse in septicemia | |
| Interferon-<mh3><piga | Virus-infected cells | MHC class I induction, antiviral activity | |
| Interferon-<mh3><pigb | Virus-infected cells | MHC class I induction, antiviral activity | |
| Interferon-<mh3><pigg | Macrophage activation factor | T cells, LGL | MHC class II induction; macrophage activation; augments NK activity; B-cell proliferation |
| Migration inhibition factor | T cells | Inhibition of migration of macrophages from site of inflammation | |
| Colony-stimulating factors (CSF) | Monocytes, macrophages, T cells | Stimulate growth & differentiation of bone marrow progenitor cells fordneutrophils | |
| G-CSF | (G-CSF); monocytesd(M-CSF); andneutrophils, monocytes, eosinophils,erythrocytes &dmegakaryocytes (GM-CSF) | ||
| M-CSF | |||
| GM-CSF |
Interleukin-1 is produced primarily by macrophages but also by LGL and B-cells. IL-1 induces helper T-cell activation, lymphokine release, and IL-2 expression, as well as B-cell proliferation and differentiation (lymphocyte activating factor). IL-1 promotes macrophage chemotaxis and cytocidal activity, polymorphonuclear leukocyte (PMN) activation and chemotaxis, and increased NK activity. IL-1 induces fever and has been called leukocytic pyrogen. The tumor necrosis factors share many of the actions of IL-1.
Interleukin-2 is produced by T cells and plays a key role in T-cell and B-cell proliferation and maturation. It enhances cytotoxic T-cell activity as well as NK and LAK activity.
Interleukin-3 is produced by T cells and stimulates differentiation of hematopoietic precursors of multiple cell lineages; it is also called multi-colonystimulating factor (multi-CSF). IL-4 is produced by activated T cells and induces B- and T-cell proliferation. IL-4 increases IgE secretion by B cells. IL-5 promotes B-cell and eosinophil growth and differentiation. IL-3, -4, and -5 play a role in allergic and antiparasitic inflammatory responses.9 IL-6 is produced by T and B cells and macrophages and fosters B-cell growth and differentiation. It also induces production of acute-phase proteins by hepatocytes. IL-7 produced by bone marrow stromal cells promotes growth of B- and T-cell precursors. IL-8 is chemotactic and may be involved in adhesion of PMNs to vascular endothelium.
The tumor necrosis factors (TNF) are produced by lymphocytes and macrophages and result in activation of macrophages, granulocytes, and cytotoxic cells. TNF enhances MHC expression and antigen presentation. TNF promotes the tumoricidal activity of monocytes and protection against microorganisms. TNF may mediate the circulatory collapse and tissue necrosis seen in septicemia.
Interferon-α and -ß promote MHC class I induction and antiviral protection. IFN-α and IFN-ß are produced by virus-infected cells early in infection and induce resistance to virus in noninfected cells. IFN-γ (macrophage activation factor) is released by T cells and LGL. It has the ability to induce class II receptor expression on many cell types (e.g., vascular endothelial cells) that do not usually express HLA-DR molecules. This promotes the participation of these cells in the immune process. IFN-γ also stimulates macrophage activation, NK activity, and B-cell proliferation.
Migration inhibition factor is produced by T cells and has the effect of keeping macrophages at the site of inflammation. Colony-stimulating factors (e.g., M-CSF, G-CSF, and GM-CSF) stimulate stem cell division and differentiation.
HUMORAL IMMUNITY
Humoral immunity (i.e., antibody-mediated immunity) involves production of antibodies locally (e.g., IgA) or systemically, triggered by antigen stimulation (antigen = antibody generator). The basic function of antibody is to bind antigen. Antigenic targets include extracellular bacteria and viruses, allergens, toxins and other foreign proteins, viral antigens expressed on infected cells, and altered autologous cellular or soluble proteins or carbohydrates. Immunoglobulins play a role in opsonization, immune complex formation, complement activation, antibody-dependent cellmediated cytotoxicity, type I hypersensitivity, and autoimmunity.
The phenomenon of change from IgM to IgG while maintaining the same specificity for antigen that occurs in the course of an immune response is known as isotype switching, an event under T-cell control. Free serum antibodies provide short-term specific immunity with a half-life of about 25 days; they are essentially absent after 1 year.
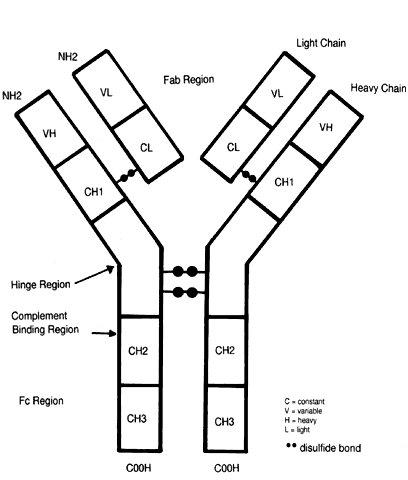
Structure
Each immunoglobulin molecule consists of two identical heavy chains and two identical light chains joined by disulfide bonds (Fig. 6). The type of heavy chain (alpha, delta, gamma, mu, epsilon) determines the immunoglobulin isotype or class (i.e., IgA, IgD, IgG, IgM, IgE, respectively). Light chains are of two types, kappa and lambda. IgG can be divided into four subclasses, IgG1, IgG2, IgG3, and IgG4. IgA has two subclasses, IgA1, IgA2. In each case, the differentiation is on the basis of specific antigenic determinants on the heavy chains. Each differentiated B cell/plasma cell produces only one type of antibody with identical heavy and light chains.
|
It should be noted that numerous molecules involved in the immune processes possess similarity in amino acid structure and sequence to immunoglobulin heavy and light chains (e.g., HLA, CD2, CD4, CD8, TCR), hence the concept of the immunoglobulin supergene family.
Each heavy and light chain is made up of constant (C) and variable (V) regions (domains). Light chains have one V and one C region, VL and CL, respectively. Heavy chains have one variable (VH) region and three or four constant (CH) regions, depending on the immunoglobulin class. Constant regions share the same structure for each immunoglobulin molecule of the same isotype and subclass.
Constant regions are involved in IgM and IgG binding to complement or IgG binding to Fc receptors of macrophages, LGL, B cells, neutrophils, and eosinophils. The Fc (Fc = crystallizable fragment) region of the immunoglobulin molecule is the CH region at the carboxyterminus end. Variable regions (both VL and VH) are found at the amino terminal end of the molecule. The variable regions are involved in binding to antigen, being known as the Fab (ab = antigen binding) region of the molecule (see Fc above). There are two antigen-binding sites per monomer. Within the VL and VH regions are hypervariable regions whose amino acid sequence is unique to that immunoglobulin molecule and specific for binding to a particular antigen. This constitutes the antigen-binding site.
Each antibody has unique antigenic determinants in its variable regions called idiotopes, the sum of which determines its idiotype. A second set of antibodies is generated that is directed at the idiotype; these are called anti-idiotype antibodies. These can regulate the response to the initial antigen by either suppressing or stimulating production of the initial antibody. Anti-anti-idiotype antibodies can also form.
Immunoglobulin Classes
IGG.
IgG is a monomer and makes up 75% of total serum immunoglobulin. It is distributed equally between the intravascular and extravascular pools. Four subclasses exist, IgG1, G2, G3, and G4, in decreasing order of frequency. IgG is the main immunoglobulin produced on second challenge to antigen. IgG binds to Fc receptors of macrophages, neutrophils, eosinophils, LGL, and B cells and can act as an opsonin. IgG (IgG1, IgG2, and IgG3, but not IgG4) is able to activate complement via its CH2 domain. Blocking antibodies are of the IgG type. It is the only immunoglobulin that can cross the placental barrier.
IGM.
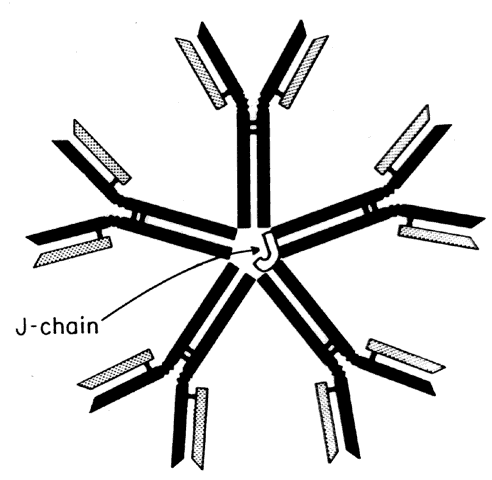
IgM is primarily intravascular in location and makes up about 10% of the immunoglobulin pool. In the circulation, IgM antibodies are found as a star-shaped pentamer consisting of five IgM monomers joined by a molecule known as the J-chain (Fig. 7). As a monomer, IgM is the major antigen receptor expressed on the surface of the mature B cell. It is the main immunoglobulin produced in the primary response to antigen. IgM is an effective agglutinator of particulate antigens. It is a potent activator of complement (binds C1 via CH4). IgM antibodies are important in defense against bacterial infections. IgM is important in the etiology of autoimmune disease because of its role in immune complex formation (e.g., rheumatoid factor is an IgM against IgG). Its large size prevents diffusion or transport across membranes (e.g., placenta).
|
IGA.
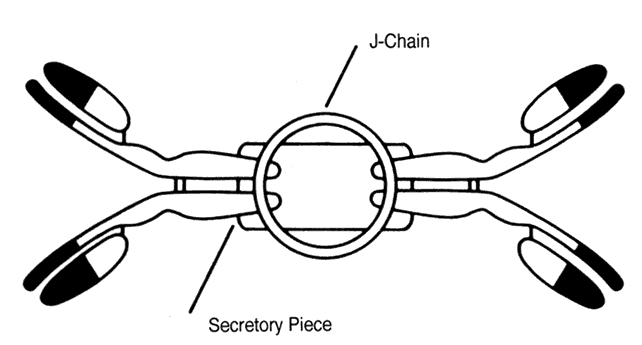
IgA constitutes 15% of total serum immunoglobulin. About 80% exists in a monomeric form, but a dimeric form exists. It is the major class of immunoglobulin in secretions such as tears, saliva, milk, and those of the respiratory and gastrointestinal (GI) tract (secretory IgA). Secretory IgA is a dimer consisting of two IgA monomers joined by a J-chain and associated with a glycoprotein called the secretory piece (Fig. 8). The secretory piece may facilitate the transport of IgA through mucosal surfaces and make the IgA molecule more resistant to degradation by proteolytic enzymes present in external secretions. There are two IgA subtypes; IgA1 is found mostly in serum, whereas IgA2 is found more in secretions. IgA fixes complement via the alternative pathway. IgA possesses potent antiviral activity in man by prevention of viral binding to respiratory and GI epithelial cells. Secretory IgA is the main immunoglobulin in the tear film, and IgA is found regularly in the superficial conjunctival epithelium in various types of conjunctivitis.10
|
IGD.
IgD is found in minimal amounts in serum. Like IgM, it acts as a major receptor for antigen on the surface of B cells.
IGE.
IgE is present in low amounts in serum. It binds to the surface of basophils and mast cells via its Fc region and acts as an antigen-binding site. Cross-linking of surface IgE by antigen promotes mast cell mediator release (type I hypersensitivity reaction). IgE plays a major role in allergy and immunity to helminths.
COMPLEMENT
The complement system comprises a cascade of about 25 soluble serum proteins. Each acts as asubstrate for the preceding protein (enzyme) in the cascade, then acts as an enzyme on the next protein in the sequence. There are two major complement pathways, the classic and alternative pathways.
In the classic pathway, which is the more rapid and efficient of the two pathways, IgG- and IgM-containing antigen-antibody complexes activate C1, causing a cascade of activation in the following order: C4, C2, C3, C5, C6, C7, C8, and C9 (Fig. 9). The alternative pathway is a slower system in which C3 is activated by microbial products (e.g., endotoxin), properdin, and/or IgA- or IgD-containing immune complexes. With C3 activation, the cascade continues in a manner similar to that of the classic system.
|
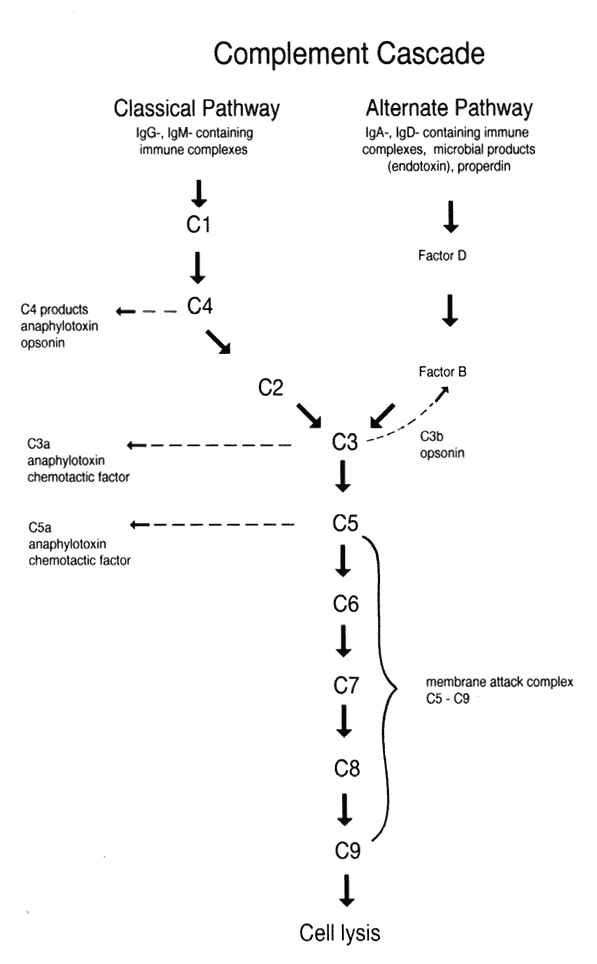
The functions of the complement system include opsonization, chemotaxis, anaphylotoxin activity, and cytotoxicity. Opsonization refers to the process of binding by C3b (or antibody) to microorganisms, facilitating phagocytosis by macrophages and neutrophils with C3b receptors (or Fc receptors in the case of opsonization by antibody). Neutrophil chemotaxis is promoted by C5a. C3a, C4a, and C5a can bind to receptors on mast cells and basophils, promoting degranulation and release of histamine and other inflammatory mediators. The cytotoxic function of complement is performed by the membrane attack complex (C5b–C9). This complex forms transmembrane channels in target cells, leading to osmotic lysis.
CLASSIC HYPERSENSITIVITY REACTIONS
The Gell-Coombs classification of hypersensitivity reactions11 has provided an organized approach to understanding the major mechanisms of immune response (Fig. 10). However, seldom does any one of the reactions occur purely by itself. Most often, more than one mechanism is at work in any immune reaction, and there is considerable interaction and overlap.
|
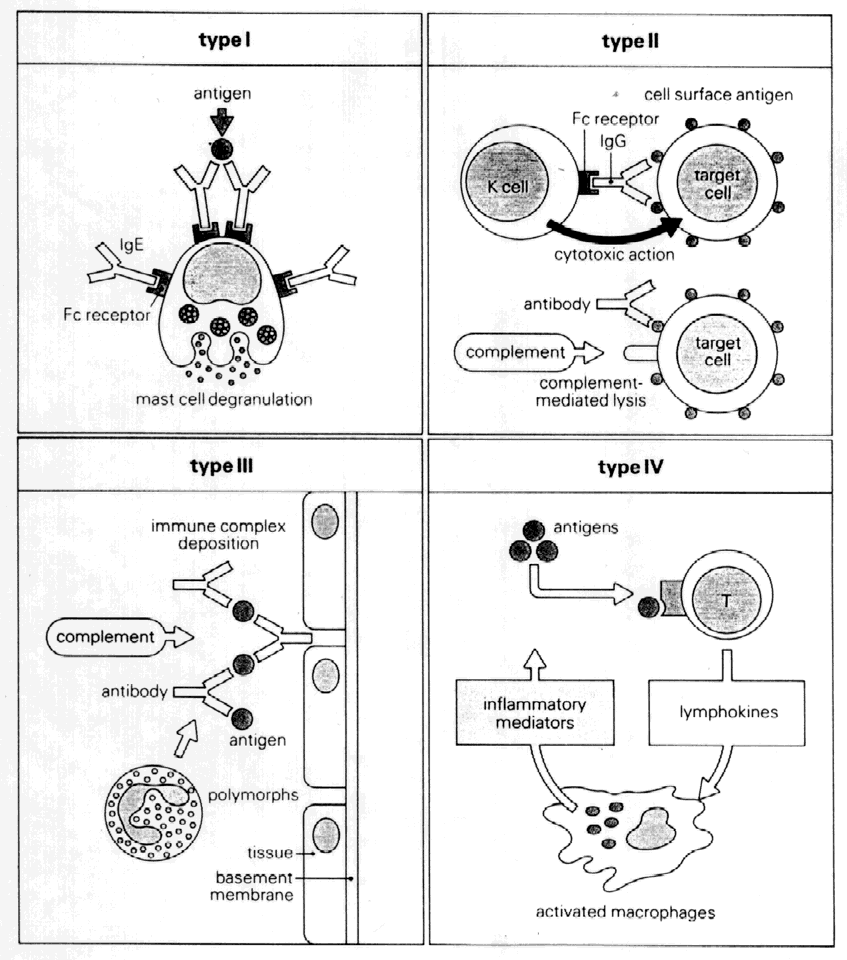
Type I: Immediate
Hypersensitivity (Anaphylaxis)
Type I hypersensitivity involves antigen binding to IgE bound to the surface of mast cells and subsequent cross-linking. This stimulates mast cell degranulation and release of preformed mediators (e.g., histamine). In addition, there is de novo synthesis of inflammatory mediators such as prostaglandins, leukotrienes, and PAF. The result is smooth muscle contraction, increased vascular permeability, and vasodilation. This is the predominant mechanism of allergic reactions and is discussed more fully elsewhere (see Mast Cells, and Allergic Conjunctivitis).
Type II: Antibody-Dependent Cytotoxicity
In type II reactions, complement-fixing antibody (e.g., IgM, IgG1, IgG2, IgG3) binds to the target cell. Complement then binds to this antibody, activating the classic pathway. The result is cell lysis. This is the pathogenic mechanism seen in Goodpasture's syndrome and in hemolysis in transfusion reactions. Pemphigoid, pemphigus, Mooren's ulcer, and thyroid-related orbitopathy are considered examples. Antibody-dependent cell-mediated cytotoxicity (i.e., K-cell activity) has also been categorized under type II reactions, although there is an obvious overlap with cell-mediated immunity.
Type III: Immune Complex-Mediated Hypersensitivity (Arthus Reaction)
Low levels of immune complexes are found in normal individuals as a result of antibody binding to antigen; these complexes are cleared by cells of the reticuloendothelial system. This process represents an effective mechanism of normal host defense. However, depending on certain factors (e.g., size of the immune complex, immunoglobulin class, antigen characteristics, hemodynamic turbulence), immune complexes can be deposited in tissues such as vascular walls and renal glomeruli. If these complexes contain IgM or IgG1–3, the complement cascade can be activated with release of chemotactic factors, neutrophil accumulation, and subsequent degranulation and enzyme release, causing local tissue damage. This mechanism is responsible for vasculitis and glomerulonephritis. Corneal immune ring formation is the result of immune complex deposition. Immune complex deposition was classically considered a major player in noninfectious uveitis; however, now its role is less clear. For example, Behçet's disease was thought to be the prototype of immune complex-mediated disease, yet T-cell-mediated mechanisms are also involved.12
Type IV: Cell-Mediated Hypersensitivity (Delayed Hypersensitivity)
Type IV hypersensitivity was initially termed delayed hypersensitivity to describe those reactions that took more than 12 hours to develop. These reactions were found to be transferable to nonimmunized hosts by lymphoid cells but not serum, indicating the cell-mediated nature of this type of immune response. Classically, four types of type IV delayed hypersensitivity reactions have been described, based on characteristic skin reactions to antigen. These types include the Jones-Mote reaction, contact hypersensitivity, tuberculin-type hypersensitivity, and granulomatous inflammation. However, in practical terms, there is considerable overlap.
To generalize, type IV cell-mediated hypersensitivity occurs as a result of class II-restricted antigen presentation by Langerhans cells or macrophages and IL-1 production, leading to helper T-cell stimulation and lymphokine release. This triggers a complex cellular response including recruitment of B cells, cytotoxic T cells, macrophages, and, to a lesser extent, neutrophils. Mechanisms of damage include cell-mediated cytotoxicity, phagocytosis, enzymatic processes, and, in the case of persistent antigen, epithelioid and giant cell formation (i.e., granulomatous inflammation).
Type IV reaction plays a major role in virus infection, tuberculosis, leprosy, and fungal infection. Sympathetic ophthalmia and Vogt-KoyanagiHarada syndrome, corneal graft rejection, phlyctenulosis, and contact allergy are all type IV reactions.
The Jones-Mote reaction is also known as cutaneous basophil hypersensitivity. It has been described as a unique form of type IV hypersensitivity in which there is a predominance of basophils. This form of hypersensitivity peaks at 24 hours, as opposed to contact and tuberculin reactions, which peak at 48 to 72 hours, and granulomatous reactions, which take 3 to 4 weeks to develop. It plays a role in vernal keratoconjunctivitis and contact lens-related giant papillary conjunctivitis (GPC).
Type V: Stimulating Antibody
Type V hypersensitivity is an antibody-mediated reaction in which autoantibody binds to cell receptors normally occupied by another molecule. This may lead to stimulation of the target cell (e.g., Graves' disease).
AUTOIMMUNITY
Tolerance
Through its inherent specificity and through certain protective mechanisms, the immune system is able to differentiate self from nonself. Tolerance to self may be attained by removal of autoreactive T-cell clones during development (clonal deletion). Another possible mechanism is clonal anergy by which B cells are rendered unresponsive to self-antigen by exposure at an early stage of development. Other possible mechanisms include sequestration of autoantigen, absence of processing and presentation of self-antigen, receptor blockade by antigen, anti-idiotype antibodies, and activity of suppressor T cells that down-regulate potential autoreactivity.13,14 However, the safeguards of immunologic self-tolerance can malfunction, resulting in immune reactions against self (i.e., autoimmunity). Several mechanisms have been implicated.
Mechanisms of Autoimmune Disease
In normal persons, autoreactive B cells and T-effector cells and autoantigens are present; however, these are not normally active because autoreactive helper T cells are functionally absent.13 Under certain circumstances, functional autoreactive helper T cells may emerge either by dysfunction of suppressor cells or by the action of T-contrasuppressor cells (which enable anti-self helper T cells to resist suppression). Also, it is possible that in some cases autoantigen may bypass helper T cells altogether and directly stimulate T-effector cells and B cells. Also, suppressor T cells that normally prevent antibody production to self-antigen could become inactivated.
Another possible autoimmune mechanism involves inappropriate expression of class II molecules on cells that normally do not possess this cell marker. This can occur in some diseases, especially in the presence of IFN-γ. This conveys to these cells the ability to act as antigen-presenting cells promoting inappropriate presentation of self-antigen. This may result in activation of helper T cells, leading to B-cell production of autoantibodies and/or activation of cytotoxic T cells.
Cross-reactivity may play a role. Alteration of self-antigen (e.g., by drugs or viruses) may enable helper T cells to function as self-reactive T-helpers and thus to generate an autoimmune response. Alternatively, the normal T- and B-cell response to foreign antigen that possesses a structure resembling self-antigen may produce antibodies that cross-react with those self-antigens of similar structure. This is known as molecular mimicry.
Nonspecific activation of the immune system by polyclonal activators (e.g., Epstein-Barr virus or bacterial lipopolysaccharides) may overwhelm normal immunoregulatory mechanisms and promote autoimmune disease. Anti-idiotype antibodies may normally play a role in controlling the immune response by binding the idiotype portion of surface immunoglobulin on B cells and suppressing B-cell antibody production. Dysfunction of anti-idiotype antibodies could lead to B-cell hyperactivity and could play a role in autoimmune disease.
Some autoantibodies play a role in autoimmune disease by their effect on surface receptors. Autoantibodies can bind to cell-surface receptors and act as agonists. An example is Graves' disease, in which an autoantibody (long-acting thyroid stimulator [LATS]) binds to the thyroid-stimulating hormone (TSH) receptor, delivering the same stimulus as TSH and driving thyroid hormone production. Other autoantibodies can block cell-surface receptors, preventing binding of hormones or other molecules to their appropriate receptors and resulting in dysfunction. An example is myasthenia gravis, in which autoantibodies bind to acetylcholine receptors on the motor endplate, resulting in defective neuromuscular transmission.
Genetic factors play a role in autoimmunity, as there are HLA associations with certain autoimmune diseases (e.g., Graves' disease and HLA-B8, -DRw3 in whites; ankylosing spondylitis and HLA-B27).
Tissue injury in autoimmune disease may occur by a number of mechanisms. Type II reactions can be involved. Circulating autoantibodies may react with self-antigens on the cell surface, triggering complement activation and cell lysis via the membrane attack complex. Also, autoantibody binding can lead to activation of cytotoxic cells, resulting in cell lysis. A second mechanism involves type III reactions. Autoantibodies may bind to free antigen, forming circulating immune complexes that deposit in tissue, followed by complement activation and subsequent inflammation. Type IV (cell-mediated) hypersensitivity mechanisms also play a role in autoreactivity.
IMMUNOSUPPRESSIVE AGENTS
A classification of immunosuppressive agents is presented in Table 3. The mechanism of action of selected agents more commonly used in the treatment of ocular inflammatory disease is discussed.
TABLE 3. Immunosuppressive Agents
| Group | Examples |
| Corticosteroids | Hydrocortisone, prednisone |
| Alkylating agents | Nitrogen mustards, cyclophosphamide, chlorambucil, thiotepa |
| Antimetabolites | |
| Purine analogues | 6-Mercaptopurine, azathioprine |
| Pyrimidine analogues | Cytosine arabinoside, bromodeoxyuridine, 5-fluorouracil |
| Folic acid analogues<pa | Methotrexate |
| Natural products | |
| Antibiotics | Cyclosporine, mitomycin, Adriamycin, actinomycin D |
| Vinca alkaloids | Vinblastine, vincristine |
| Other | Colchicine, L-asparaginase |
| Ionizing radiation |
Corticosteroids
Corticosteroids exert their effect by binding with receptor proteins in the cytoplasm, forming a steroid-receptor complex. This complex migrates to the nucleus, where it binds to chromatin and influences RNA synthesis and ultimately the synthesis of specific proteins.15 The result is modified cell function. The anti-inflammatory and immunosuppressive effects of corticosteroids include inhibition of macrophage and neutrophil migration, as well as induction of lymphocytopenia, eosinopenia, and monocytopenia. Glucocorticoids affect inflammatory chemical mediators by inhibiting degranulation of neutrophils, mast cells, and basophils; stabilizing lysosomes; and suppressing the action of lymphokines. Corticosteroids reduce capillary permeability and suppress vasodilation. Corticosteroids act as inhibitors of prostaglandin and leukotriene synthesis by inhibiting the enzyme (phospholipase A2) responsible for production of arachidonic acid from cell membrane phospholipid.16 Unlike cytotoxic agents, corticosteroids do not cause lysis of immune cells in humans.
Cytotoxic Agents
Cytotoxic agents include alkylating agents and antimetabolites such as purine analogues (6-mercaptopurine, azathioprine), pyrimidine analogues, and the folic acid analogue methotrexate. As a group, these compounds interfere with the synthesis of nucleic acids and proteins. Any rapidly proliferating cells (e.g., lymphocytes involved in an immune response) are extremely sensitive to such agents. Although interference with nucleic acid and protein synthesis has classically been considered the major mode of action of these agents, other mechanisms are undoubtedly involved.
ALKYLATING AGENTS.
Alkylating agents include nitrogen mustard and its derivatives cyclophosphamide and chlorambucil. Cyclophosphamide must be metabolized in the liver before becoming active, whereas chlorambucil can act directly. Alkylating agents cause covalent links (alkylation) with various nucleophilic substances, such as phosphate, amino, sulfhydryl, hydroxyl, carboxyl, and imidazole groups. In DNA, the key target is the 7 nitrogen of the purine base guanine. Alkylation of these guanine residues induces abnormal base pair formation between the altered guanine and thymine (rather than cytosine, as occurs normally). The result is DNA miscoding. Also, breaks in DNA strands can be induced and cross-linking of DNA strands can occur, resulting in failure of separation during division.17 Other mechanisms are also involved.
At clinical doses, these agents are cytotoxic for lymphocytes and affect B and T cells about equally. At higher doses, alkylating agents have a greater effect on B cells than on T cells.18,19 Alkylating agents are more potent immunosuppressive agents than the antimetabolites.18
ANTIMETABOLITES.
Purine Analogues. Purine analogues, including 6-mercaptopurine and azathioprine, interfere with purine synthesis and thus alter DNA, RNA, and protein synthesis. Azathioprine is a prodrug, being converted in the liver to 6-mercaptopurine. This is further converted to thioinosinic acid and other metabolites. These metabolites inhibit enzymes involved in DNA synthesis and are also incorporated into nucleic acids, producing abnormal base sequences. The effect of these antimetabolites probably involves other mechanisms as well.17
The major effect of purine analogues at usual doses appears to be on T cells, but at higher doses, B cells are affected.19,20
Methotrexate.
This antimetabolite is a potent inhibitor of dihydrofolate reductase, which catalyzes the reduction of dihydrofolate to tetrahydrofolate. Inhibition of this process by methotrexate limits the production of thymidylate, which is essential for DNA synthesis and cell division. Thus, methotrexate produces an antiproliferative effect, which explains, at least in part, its immunosuppressive effect.21 Both B- and T-cell functions are suppressed.20
Cyclosporine
This lipid-soluble cyclic peptide containing 11 amino acid residues is a metabolite of the fungus Tolypocladium inflatum Gams. The major effect of cyclosporine is inhibition of the early stage of T-cell activation by primarily reducing IL-2 production, although this is probably only one of several mechanisms. Cyclosporine diffuses through the plasma membrane of the T lymphocyte, where it binds to the cytoplasmic protein cyclophilin. This enzyme plays a role in a series of steps that lead to stimulation of specific gene transcription, including that for IL-2.22 By this method, cyclosporine causes a selective inhibitory effect on the synthesis of IL-2 by helper T cells and thus an inhibitory effect on T-helper lymphocyte activation. Production of lymphokines other than IL-2 (IL-1, IL-3, MAF, MIF, IFN-γ) is also affected. Cyclosporine may inhibit expression of IL-2 receptors on the surface of T lymphocytes and may interfere with T-cell surface receptors that recognize HLA-DR antigens on antigen-presenting cells.20 Cyclosporine also inhibits induction and proliferation of cytotoxic T lymphocytes.23 The overall result is suppression of cell-mediated immunity. Cyclosporine also may suppress humoral immunity through inhibition of T- and B-cell cooperation.24 Cyclosporine has also been reported to have an immunosuppressive effect by binding to prolactin receptors on the surface of lymphocytes; this prevents binding by prolactin, a known immunostimulator. Bromocriptine, a dopamine agonist that suppresses prolactin secretion, increases the effectiveness of cyclosporine.25 Cyclosporine has no effect on hematopoietic stem cells or nonlymphocytic leukocytes.
This section has been solely an overview of the mechanism of action of more commonly used immunosuppressive agents. Clinical indications, dosage, and adverse effects have not been discussed. As a group, these are potent agents, and the possibility of severe side effects (e.g., bone marrow suppression and secondary malignancies with cytotoxic agents) is great. Thus, immunosuppressives must be used selectively and cautiously.
ANATOMY/IMMUNOLOGY CORRELATION
Nonimmune Protective Mechanisms of the External Eye
The eye features several protective mechanisms that are nonimmune in nature. First of all, the eyelids and blink reflex provide a physical barrier to foreign materials reaching the eye. The tears themselves have a diluting and flushing effect. Also, the tear film plays a role in maintaining the integrity of the corneal and conjunctival epithelia, which provide a protective anatomic barrier. The normal bacterial flora of the eyelids also has a beneficial role in inhibiting growth of potential pathogens.
TEARS.
In addition to its nonimmune protective functions, the tear film provides an immunologic barrier to the environment. The tear film contains lactoferrin, lysozyme, and beta-lysin, which have antimicrobial effects. Immunoglobulins, complement, and cytokines are present. In addition, inflammatory mediators such as histamine and prostaglandins are found. Lysozyme is a cationic, low-molecular-weight enzyme that attacks the mucopeptides of the cell walls of susceptible bacteria. Lactoferrin has bacteriostatic properties by making certain metals unavailable for microbial agents.
IgA is the major immunoglobulin in tears and is found mostly in the secretory form. IgG is usually present with lesser amounts of IgM and IgE. Plasma cells found in the main and accessory lacrimal glands produce IgA in the dimeric form (two IgA monomers joined by a J-chain). This passes through the basement membrane of the acinar cells, which attach a secretory component. Secretory IgA is then released by the acinar cells into the lumen of the lacrimal ducts. IgM can bind secretory component and probably follows a similar route. It was once thought that IgG and IgE came solely from the serum, but there is evidence of local production as well. However, the exact route of entry of IgG and IgE into the tears is unknown at present.26 IgA is important in promoting microbial phagocytosis, inhibiting epithelial binding by microbials, interfering with bacterial exotoxins, and facilitating antibody-dependent cell-mediated cytotoxicity.27
CONJUNCTIVA.
The conjunctiva also provides an immunologic as well as a physical barrier to the environment. The conjunctiva contains blood vessels, lymphoid tissue, and immunoreactive cells, including lymphocytes, neutrophils, Langerhans cells, neutrophils, and mast cells. The lymphoid cell population of the conjunctiva-associated lymphoid tissue (CALT) is analogous to that of other mucosa-associated lymphoid tissues (MALT).28 In the conjunctival epithelium of normals, small numbers of lymphocytes are interspersed in the epithelial layer, mainly suppressor/cytotoxic T cells.10,29 In the substantia propria, T lymphocytes represent about one third of the cell population, with helper T cell and suppressor/cytotoxic T-cell populations in about equal proportions. In a quiet conjunctiva, most of the CD8+ cells would be expected to be suppressor cells. B cells are present in the substantia propria, but in smaller numbers, arranged in aggregates mostly in the fornices. Langerhans cells are the major antigen-presenting cells of the conjunctiva (as they are for cornea and skin) and are located in the epithelium.30 In addition, non-Langerhans dendritic cells are found in the substantia propria and basilar epithelium and also function as antigen-presenting cells.30
There are approximately 50 million mast cells in the ocular and adnexal tissues of the human eye.31 In normal conditions, mast cells are concentrated in the substantia propria of the conjunctiva but are not found in the conjunctival epithelium. However, in disease states such as vernal and contact lens- induced giant papillary conjunctivitis (GPC), mast cells infiltrate the conjunctival epithelium. Each mast cell has as many as 500,000 receptors for IgE; IgE is bound to 10% of these receptors, thus coating the mast cell surface.7 The Fab portion of the IgE molecule extends from the mast cell surface, poised to bind antigen and thus trigger mast cell degranulation, resulting in type I immune reaction. There are two main types of mast cells in the conjunctiva, defined by their protease (tryptase and chymase) content. The T mast cell is tryptase positive and chymase negative, whereas the TC mast cell is tryptase positive and chymase positive. In the normal conjunctival substantia propria, both T and TC mast cells are present, with the latter being predominant. In inflammation (e.g., vernal, GPC), the TC mast cells greatly increase in number and also are found in the epithelium.32
Scattered neutrophils are found in the epithelium and substantia propria. Eosinophils and basophils are not present in normal conjunctiva but are present in disease states.
Lymphoid cells sensitized to antigen exit via lymphatic channels to regional lymph nodes (submandibular and preauricular), then via efferent lymphatics to the thoracic duct and to the bloodstream. The sensitized lymphocytes return via the bloodstream to take up residence in conjunctival substantia propria (mostly T cells) and in lacrimal and accessory lacrimal glands (mostly B cells). Some cells probably migrate to other sites of MALT as well.
CORNEA.
The normal cornea is devoid of blood vessels, lymphatics, and inflammatory cells. Class II antigen-bearing Langerhans cells are found in corneal epithelium, with the greatest density at the periphery, decreasing toward the central cornea. Although complement components C3, C4, and C5 are found throughout the corneal stroma, there is more complement in the peripheral cornea than in the central cornea overall, especially with regard to C1. The major source of complement is the limbal vasculature, with complement components diffusing from the peripheral to the central cornea. Being the largest complement component, C1 is less likely to diffuse centrally. Immunoglobulins in the human cornea probably diffuse inward from the limbal vessels. IgG is the predominant immunoglobulin in the cornea and probably the most important in microbial defense. Both IgG and, to a lesser extent, IgA are found through the whole stroma, each distributed smoothly from peripheral to central cornea. However, IgM is found only in the peripheral cornea, probably because its large size restricts diffusion. Stromal edema associated with corneal inflammation facilitates diffusion of immunoglobulins and complement toward the central cornea from the limbal vessels, reducing differences in distribution.33
Thus, immunologic differences between the central and peripheral cornea exist. The peripheral cornea has more Langerhans cells, more IgM, and more C1 and is adjacent to conjunctival lymphatics and vasculature. This has implications in the generation of peripheral corneal disease. Antigens in the peripheral cornea are closer to the conjunctival vessels and lymphatics and thus should be able to elicit an immune response more easily; likewise, conjunctival immune responses should be more likely to affect the peripheral cornea. Because C1 is the recognition unit for the classic pathway of complement activation, the complement cascade is more likely to be triggered in the peripheral cornea. The increased amount of IgM in the peripheral cornea may play a role in peripheral corneal rheumatoid melts, which is consistent with the fact that rheumatoid factor is an IgM antibody directed against IgG.33
The immune privilege of the cornea is related to several factors, including the lack of blood vessels and lymphatics in the cornea and the fact that the central cornea is relatively free of C1, IgM, and Langerhans cells. Anterior chamber-associated immune deviation (ACAID) also plays a role.34 This term describes the phenomenon of down-regulation of cell-mediated immunity in the face of antigen exposure, which is unique to the anterior chamber. Production of transforming growth factor-ß (TGF-ß) by cells in the iris and ciliary body seems to be an integral component of this phenomenon.12 Also, classical antigen-presenting cells (MHC class II) are not normally found in the anterior chamber of the eye.