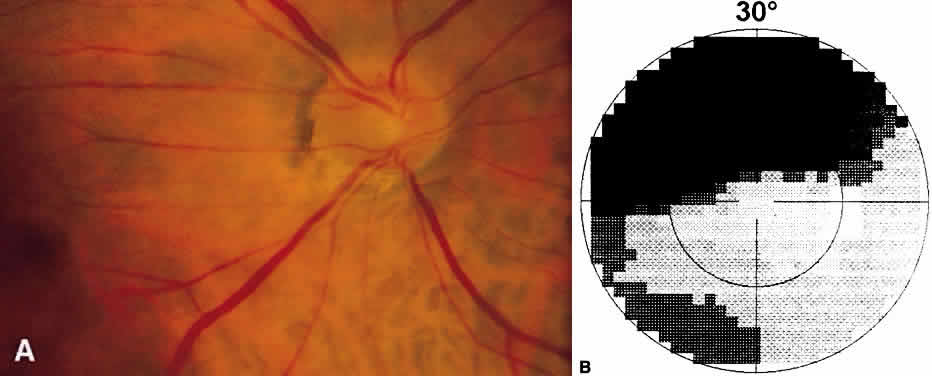
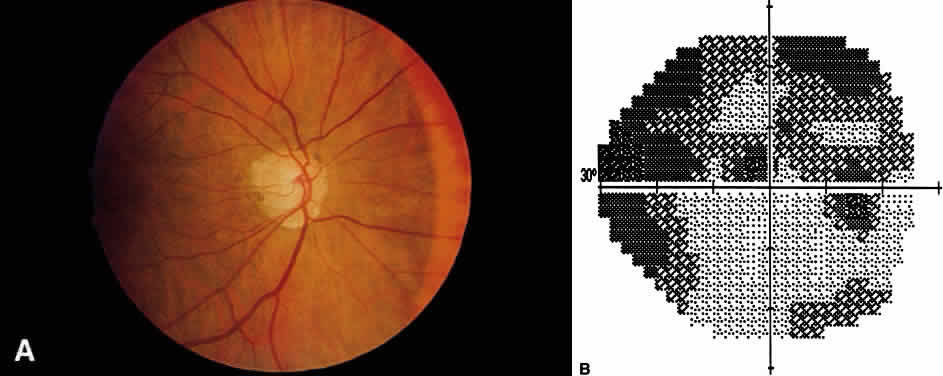
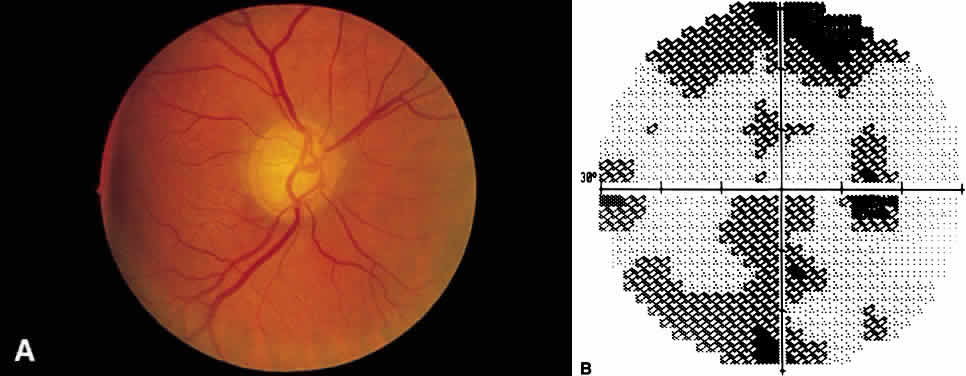
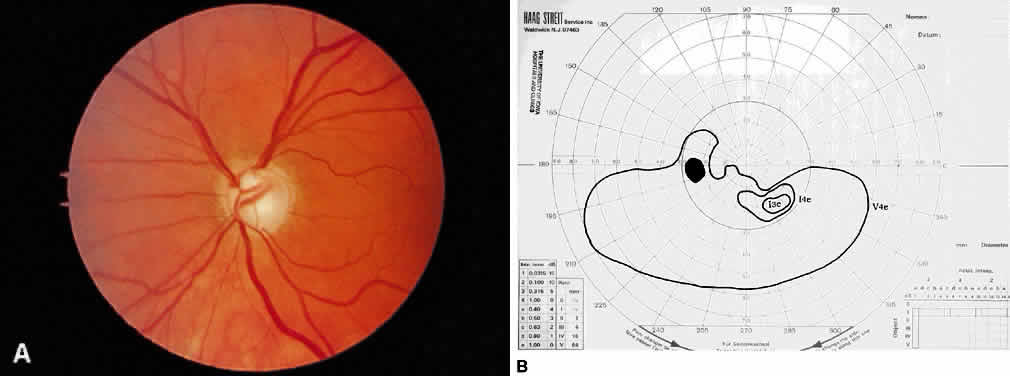
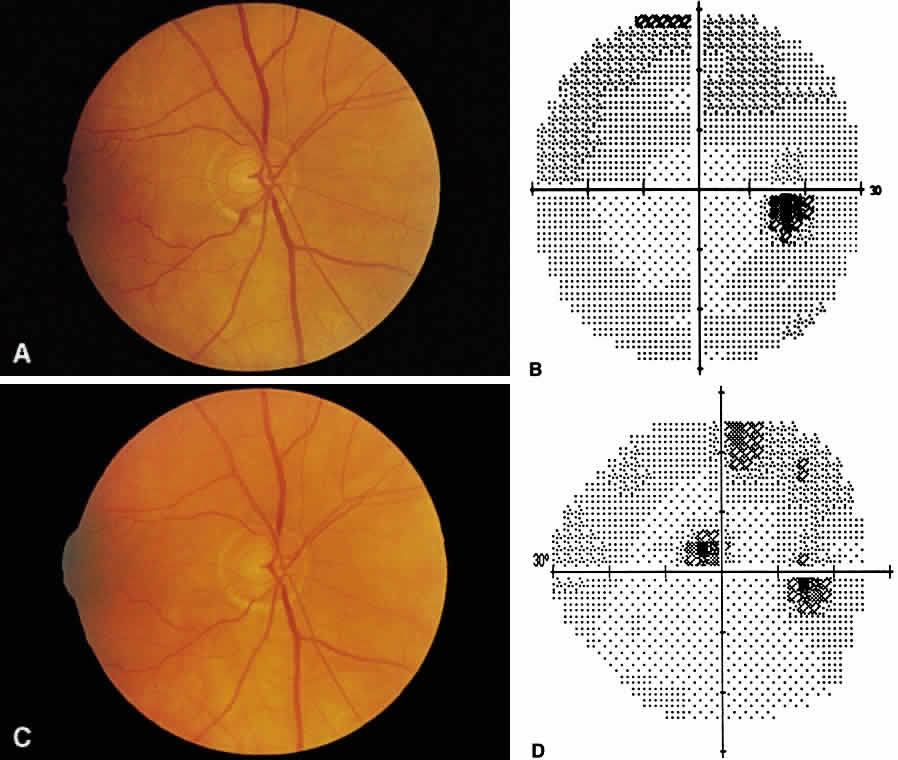
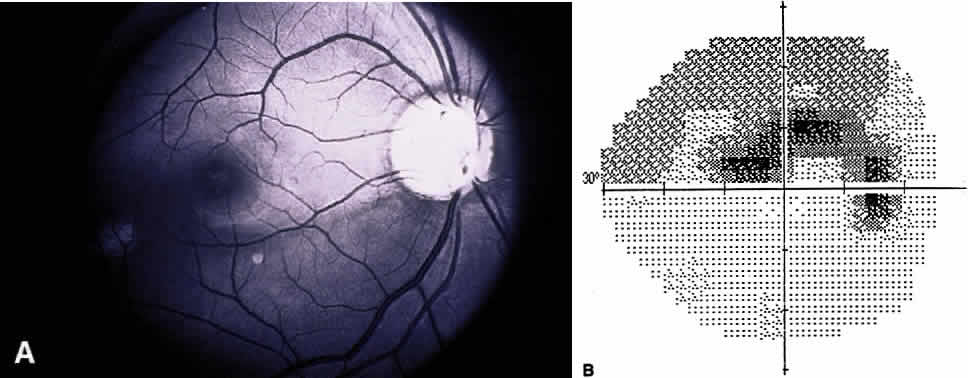
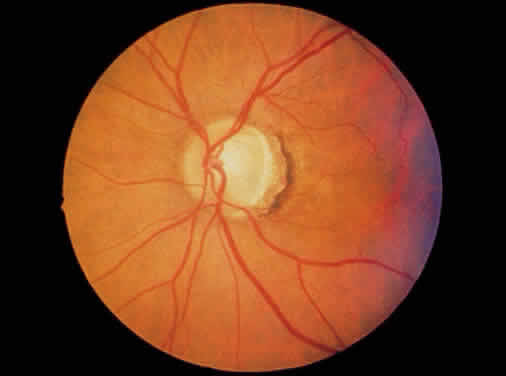
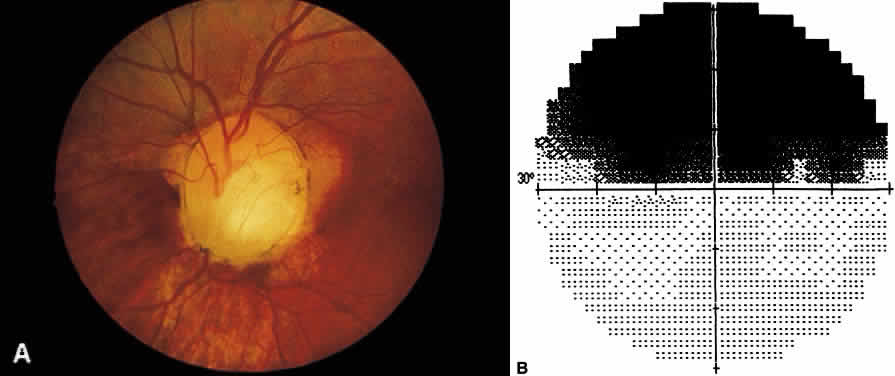
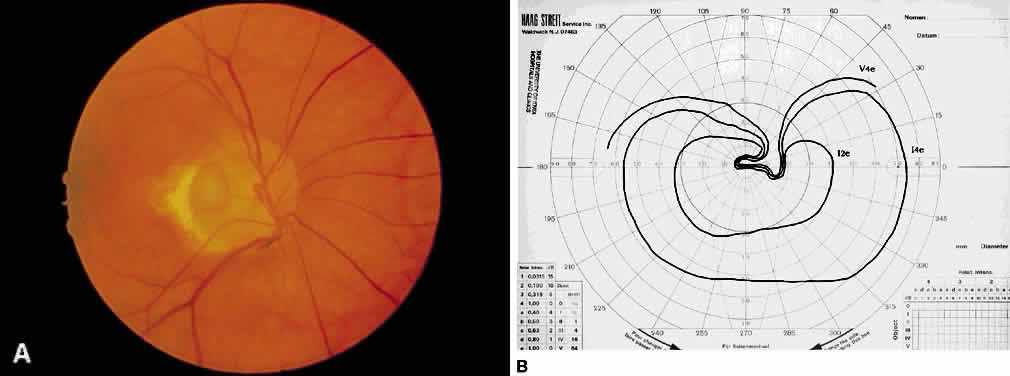
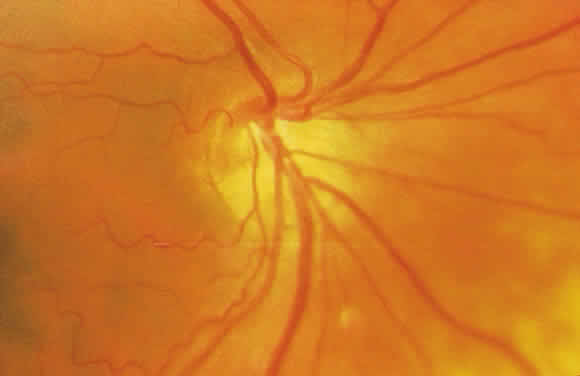
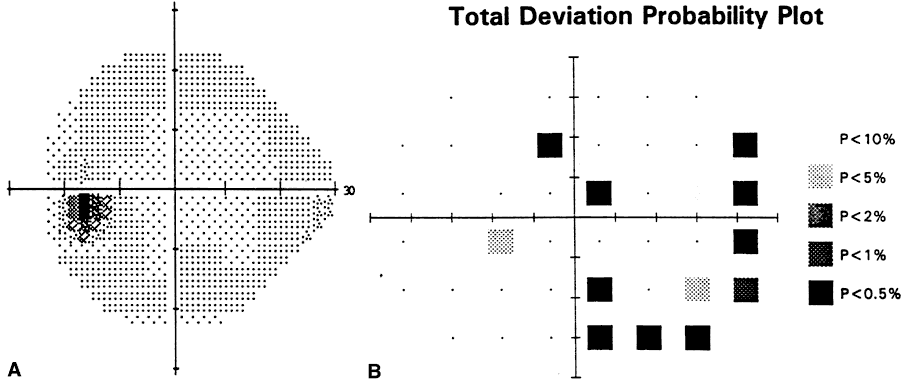
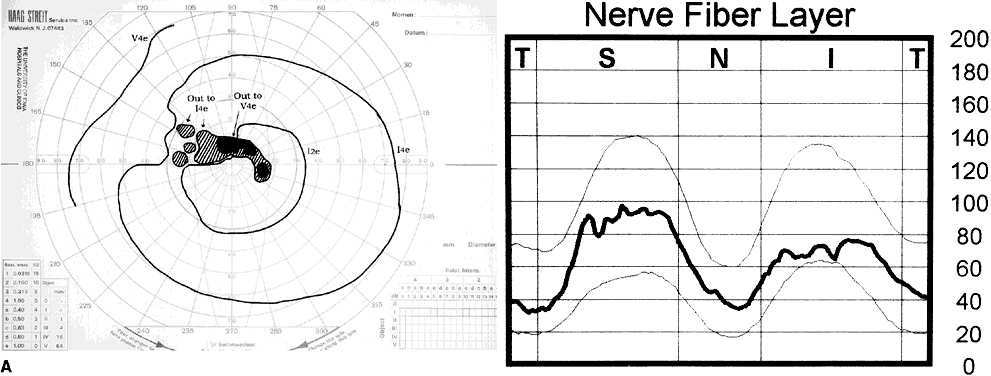
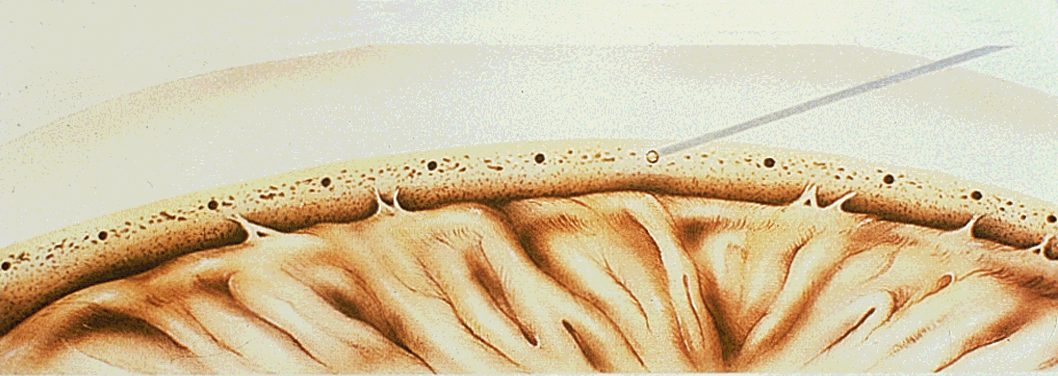
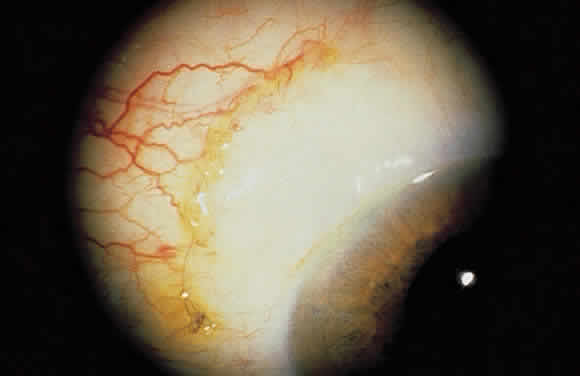
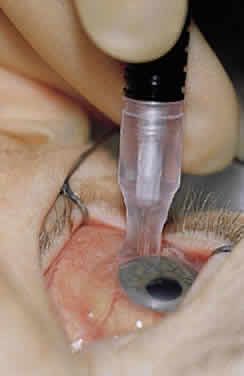
1. Leydhecker W, Akiyama K, Neumann HG: Der intraokulare Druck gesunder menschlicher Augen. Klin Monatsbl Augenheilkd 133:662, 1958 2. Jay JL, Murdoch JR: The rate of visual field loss in untreated primary open angle glaucoma. Br J Ophthalmol 77:176, 1993 3. Caprioli J, Spaeth GL: Comparison of visual field defects in the low-tension glaucomas with those
in the high-tension glaucomas. Am J Ophthalmol 97:730, 1984 4. Chauhan B, Drance SM: The influence of intraocular pressure on visual field damage in patients
with normal tension and high tension glaucoma. Arch Ophthalmol 103:1145, 1990 5. Kass MA: Normal-pressure glaucoma. Am J Ophthalmol 125:242, 1998 6. Sommer AE, Tielsch JM, Katz J et al: Relationship between intraocular pressure and primary open angle glaucoma
among white and black Americans. Arch Ophthalmol 109: 1090, 1991 7. Grant WM: Further studies on facility of flow through the trabecular meshwork. Arch Ophthalmol 60:523, 1958 8. Li Y, Yi Y: Histochemical and electron microscopic studies of the trabecular meshwork
in primary open-angle glaucoma. Eye Sci 1:17, 1985 9. Fechtner RD, Weinreb RN: Mechanisms of optic nerve damage in primary open angle glaucoma. Surv Ophthalmol 39:23, 1994 10. Nickells RW: Retinal ganglion cell death in glaucoma: The how, the why and the maybe. J Glaucoma 5:345, 1996 11. Ashton N: The exit pathway of the aqueous. Trans Ophthalmol Soc UK 80:397, 1960 12. Speakman JS, Leeson TS: Site of obstruction to aqueous outflow in chronic simple glaucoma. Br J Ophthalmol 46:321, 1962 13. Knepper PA, Covici S, Fadel JR et al: Surface-tension properties of hyaluronic acid. J Glaucoma 4:194, 1995 14. Rodrigues MM, Spaeth GL, Sivalingam E et al: Value of trabeculectomy specimens in glaucoma. Ophthalmic Surg 9(2):29, 1978 15. Segawa K: Electron microscopic studies of the trabecular meshwork in primary open-angle
glaucoma. Ann Ophthalmol 11:49, 1979 16. Alvarado JA, Yun AJ, Murphy CG: Juxtacanalicular tissue in primary open angle glaucoma and in nonglaucomatous
normals. Arch Ophthalmol 104:1517, 1986 17. Rohen JW: Presence of matrix vesicles in the trabecular meshwork of glaucomatous
eyes. Graefes Arch Clin Exp Ophthalmol 218:171, 1982 18. Rohen JW: Why is intraocular pressure elevated in chronic simple glaucoma? Ophthalmology 90:758, 1983 19. Babizhayev MA, Brodskaya MW: Fibronectin detection in drainage outflow system of human eyes in ageing
and progression of open-angle glaucoma. Mech Ageing Dev 47:145, 1989 20. Umihira J, Nagata S, Nohara M et al: Localization of elastin in the normal and glaucomatous human trabecular
meshwork. Invest Ophthalmol Vis Sci 35:486, 1994 21. Fine BS, Yanoff M, Stone RA: A clinicopathologic study of four cases of primary open-angle glaucoma
compared to normal eyes. Am J Ophthalmol 91:88, 1981 22. Nesterov AP, Batmanov YE: Trabecular wall of Schlemm's canal in the early stage of primary open-angle
glaucoma. Am J Ophthalmol 78:639, 1974 23. Moses RA, Grodski WJ Jr, Etheridge EL et al: Schlemm's canal: the effect of intraocular pressure. Invest Ophthalmol Vis Sci 20:61, 1981 24. Buller C, Johnson D: Segmental variability of the trabecular meshwork in normal and glaucomatous
eyes. Invest Ophthalmol Vis Sci 35:3841, 1994 25. Tripathi RC, Li J, Chan WFA et al: Aqueous humor in glaucomatous eyes contains an increased level of TGF-β2. Exp Eye Res 59:723, 1994 26. González-Avila G, Ginebra M, Hayakawa T et al: Collagen metabolism in human aqueous humor from primary open-angle glaucoma. Decreased
degradation and increased biosynthesis play a role in
its pathogenesis. Arch Ophthalmol 113:1319, 1995 27. Stone EM, Fingert JH, Alward WLM et al: Identification of a gene that causes primary open angle glaucoma. Science 275:668, 1997 28. Alward WLM, Fingert JH, Johnson AT et al: The phenotype of primary open angle glaucoma patients with mutations in
the GLC1A gene. Invest Ophthalmol Vis Sci Abstr 38:4335, 1997 29. Levy NS, Crapps EE, Bonney RC: Displacement of the optic nerve head. Response to acute intraocular pressure
in primate eyes. Arch Ophthalmol 99:2166, 1981 30. Zeimer RC, Ogura Y: The relation between glaucomatous damage and optic nerve head mechanical
compliance. Arch Ophthalmol 107:1232, 1989 31. Coleman AL, Quigley HA, Vitale S et al: Displacement of the optic nerve head by acute changes in intraocular pressure
in monkey eyes. Ophthalmology 98:35, 1991 32. Quigley HA, Hohman RM, Addicks EM et al: Morphologic changes in the lamina cribrosa correlated with neural loss
in open-angle glaucoma. Am J Ophthalmol 95:673, 1983 33. Miller KN, Quigley HA: The clinical appearance of the lamina cribrosa as a function of the extent
of glaucomatous optic nerve damage. Ophthalmology 95:135, 1988 34. Morrison JC, Dorman-Peace ME, Dunkelberger GR et al: Optic nerve head extracellular matrix in primary optic atrophy and experimental
glaucoma. Arch Ophthalmol 108:1020, 1990 35. Hernandez MR, Andrzejewska WM, Neufeld AH: Changes in the extracellular matrix of the human optic nerve head in primary
open-angle glaucoma. Am J Ophthalmol 109: 180, 1990 36. Hernandez MR: Ultrastructural immunocytochemical analysis of elastin in the human lamina
cribrosa. Changes in elastic fibers in primary open-angle glaucoma. Invest Ophthalmol Vis Sci 33:2891, 1992 37. Quigley HA, Brown A, Dorman-Pease ME: Alterations in elastin of the optic nerve head in human and experimental
glaucoma. Br J Ophthalmol 75:552, 1991 38. Minckler DS, Bunt AH, Johanson GW: Orthograde and retrograde axoplasmic transport during acute ocular hypertension
in the monkey. Invest Ophthalmol Vis Sci 16: 426, 1977 39. Minckler DS, Bunt AH, Klock IB: Radioautographic and cytochemical ultrastructural studies of axoplasmic
transport in the monkey optic nerve head. Invest Ophthalmol Vis Sci 17:33, 1978 40. Quigley HA, Anderson DR: The dynamics and location of axonal transport blockade by acute intraocular
pressure elevation in primate optic nerve. Invest Ophthalmol 15: 606, 1976 41. Radius RL, Bade B: Pressure-induced optic nerve axonal transport interruption in cat eyes. Arch Ophthalmol 99: 2163, 1981 42. Pearson HE, Stoffler DJ: Retinal ganglion cell degeneration following loss of postsynaptic target
neurons in the dorsal lateral geniculate nucleus of the adult cat. Exp Neurol 116:163, 1992 43. Schultz M, Raju T, Ralston G et al: A retinal ganglion cell neurotrophic factor purified from the superior
colliculus. J Neurochem 55:832, 1990 44. Hayreh SS: Pathogenesis of optic nerve head changes in glaucoma. Semin Ophthalmol 1:1, 1986 45. Hayreh SS: Structure and blood supply of the optic nerve. In Heilmann K, Richardson
KT (eds): Glaucoma: Conceptions of a Disease, p 78. Stuttgart, Thieme, 1978 46. François J, Neetens A: Vascularity of the eye and the optic nerve in glaucoma. Arch Ophthalmol 71:219, 1964 47. Quigley HA, Hohman RM, Addicks EM et al: Blood vessels of the glaucomatous optic disc in experimental primate and
human eyes. Invest Ophthalmol Vis Sci 25:918, 1984 48. Hayreh SS: Progress in the understanding of the vascular etiology of glaucoma. Curr Opin Ophthalmol 5:11, 1994 49. Hayreh SS, Zimmerman MB, Podhajsky P et al: Nocturnal arterial hypotension and its role in optic nerve head and ocular
ischemic disorders. Am J Ophthal 117:603, 1994 50. Dielemans I, de Jong PTVM, Stolk R et al: Primary open-angle glaucoma, intraocular pressure, and diabetes mellitus
in the general elderly population. The Rotterdam Study. Ophthalmology 103:1271, 1996 51. Kahn HA, Leibowitz HM, Ganley JP et al: The Framingham Eye Study. I. Outline and major prevalence findings. Am J Epidemiol 106:17, 1977 52. Tielsch JM, Katz J, Quigley HA et al: Diabetes, intraocular pressure, and primary open-angle glaucoma in the
Baltimore Eye Survey. Ophthalmology 102:48, 1995 53. Pillunat LE, Stodtmeister R, Wilmanns I et al: Autoregulation of ocular blood flow during changes in intraocular pressure: Preliminary
results. Graefes Arch Clin Exp Ophthalmol 223:219, 1985 54. Robert Y, Steiner D, Hendrickson P: Papillary circulation dynamics in glaucoma. Graefes Arch Clin Exp Ophthalmol 227:436, 1989 55. Quigley HA, Nickells RW, Kerrigan LA et al: Retinal ganglion cell death in experimental glaucoma and after axotomy
occurs by apoptosis. Invest Ophthalmol Vis Sci 36:774, 1995 56. Büchi ER: Cell death in the rat retina after a pressure-induced ischaemia-reperfusion
insult: An electron microscopic study. I. Ganglion cell layer and
inner nuclear layer. Exp Eye Res 55:605, 1992 57. Garcia-Valenzuela E, Shareef S, Walsh J et al: Programmed cell death of retinal ganglion cells during experimental glaucoma. Exp Eye Res 61:33, 1995 58. Radius RL, Anderson DR: Rapid axonal transport in primate optic nerve. Arch Ophthalmol 99:650, 1981 59. Dandona L, Hendrickson A, Quigley HA: Selective effects of experimental glaucoma on axonal transport by retinal
ganglion cells to the dorsal lateral geniculate nucleus. Invest Ophthalmol Vis Sci 32:1593, 1991 60. Choi D: Glutamate excitotoxicity and diseases of the nervous system. Neuron 1:623, 1988 61. Schumer RA, Podos SM: The nerve of glaucoma! Arch Ophthalmol 112:37, 1994 62. Caprioli J: Neuroprotection of the optic nerve in glaucoma. Acta Ophthalmol Scand 75:364, 1997 63. Dreyer EB, Zurakowski D, Schumer RA et al: Elevated glutamate levels in the vitreous body of humans and monkeys with
glaucoma. Arch Ophthalmol 114:299, 1996 64. Leske MC: The epidemiology of open-angle glaucoma: A review. Am J Epidemiol 118:166, 1983 65. Leibowitz HM, Krueger DE, Maunder LR et al: The Framingham Eye Study monograph: An ophthalmological and epidemiological
study of cataract, glaucoma, diabetic retinopathy, macular degeneration, and
visual acuity in a general population of 2631 adults, 1973-1975. Surv Ophthalmol 24(suppl):335, 1980 66. Tielsch JM, Sommer A, Katz J et al: Racial variations in the prevalence of primary open-angle glaucoma. The
Baltimore Eye Survey. JAMA 266:369, 1991 67. Sommer A, Tielsch JM, Katz J: Racial differences in the cause-specific prevalence of blindness in East
Baltimore. N Engl J Med 325:1412, 1991 68. The international bank for reconstruction and development/The World Bank: World
Development Report 1993: Investing in Health. New York, Oxford
University Press, 1993 69. Amoni SS: Pattern of presentation of glaucoma in Kaduna, Nigeria. Glaucoma 2:445, 1980 70. Hollows FC, Graham PA: Intra-ocular pressure, glaucoma, and glaucoma suspects in a defined population. Br J Ophthalmol 50:570, 1966 71. Klein BEK, Klein R, Sponsel WE et al: Prevalence of glaucoma. The Beaver Dam Eye Study. Ophthalmology 99: 1499, 1992 72. Coffey M, Reidy A, Wormald R et al: Prevalence of glaucoma in the west of Ireland. Br J Ophthalmol 77:17, 1993 73. Dielemans I, Vingerling JR, Wolfs RCW et al: The prevalence of primary open-angle glaucoma in a population-based study
in the Netherlands. The Rotterdam Study. Ophthalmology 101:1851, 1994 74. Mitchell P, Smith W, Attebo K et al: Prevalence of open-angle glaucoma in Australia. The Blue Mountains Eye
Study. Ophthalmology 103:1661, 1996 75. Mason RP, Kosoko O, Wilson MR et al: National survey of the prevalence and risk factors of glaucoma in St. Lucia, West
Indies. I. Prevalence findings. Ophthalmology 96: 1363, 1989 76. Leske MC, Connell AMS, Schachat AP et al: The Barbados Eye Study. Prevalence of open angle glaucoma. Arch Ophthalmol 112:821, 1994 77. Shiose Y, Kitazawa Y, Tsukahara S, et al: Epidemiology of glaucoma in Japan: A nationwide glaucoma survey. Jpn J Ophthalmol 35:133, 1991 78. Bengtsson B: Incidence of manifest glaucoma. Br J Ophthalmol 73:483, 1989 79. Armaly MF: Ocular pressure and visual fields. A ten-year follow-up study. Arch Ophthalmol 81:25, 1969 80. Norskov K: Routine tonometry in ophthalmic practice. II. Five-year follow-up. Acta Ophthalmol 48:873, 1970 81. Perkins ES: The Bedford glaucoma survey. I. Long-term follow-up of borderline cases. Br J Ophthalmol 57:179, 1973 82. Walker WM: Ocular hypertension. Follow-up of 109 cases from 1963 to 1974. Trans Ophthalmol Soc UK 94:525, 1974 83. Wilensky JT, Podos SM, Becker B: Prognostic indicators in ocular hypertension. Arch Ophthalmol 91:200, 1974 84. Linnér E: Ocular hypertension. I. The clinical course during ten years without therapy. Aqueous
humour dynamics. Acta Ophthalmol 54:707, 1976 85. Kitazawa Y, Horie T, Aoki S, et al: Untreated ocular hypertension. A long-term prospective study. Arch Ophthalmol 95:1180, 1977 86. David R, Livingston DG, Luntz MH: Ocular hypertension—a long-term follow-up of treated and untreated
patients. Br J Ophthalmol 61:668, 1977 87. Hart WM Jr, Yablonski M, Kass MA, et al: Multivariate analysis of the risk of glaucomatous visual field loss. Arch Ophthalmol 97:1455, 1979 88. Lundberg L, Wettrell K, Linnér E: Ocular hypertension. A prospective twenty-year follow-up study. Acta Ophthalmol 65:705, 1987 89. Quigley HA, Addicks EM: Chronic experimental glaucoma in primates. II. Effect of extended intraocular
pressure elevation on optic nerve head and axonal transport. Invest Ophthalmol Vis Sci 19:137, 1980 90. Bankes JLK, Perkins ES, Tsolakis S et al: Bedford Glaucoma Survey. Br Med J 30:791, 1968 91. Tielsch JM, Katz J, Sommer A et al: Family history and risk of primary open angle glaucoma. The Baltimore Eye
Survey. Arch Ophthalmol 112:69, 1994 92. Perkins ES, Phelps C: Open-angle glaucoma, ocular hypertension, low-tension glaucoma, and refraction. Arch Ophthalmol 100:1464, 1982 93. Wilson MR, Hertzmark E, Walker AM: A case-control study of risk factors in open-angle glaucoma. Arch Ophthalmol 105:1066, 1987 94. David R, Zangwill LM, Tessler Z et al: The correlation between intraocular pressure and refractive status. Arch Ophthalmol 103:1812, 1985 95. Daubs JG, Crick RP: Effect of refractive error on the risk of ocular hypertension and open-angle
glaucoma. Trans Ophthalmol Soc UK 101:121, 1981 96. Armstrong JR, Daily RK, Dobson HL et al: The incidence of glaucoma in diabetes mellitus. A comparison with the incidence
of glaucoma in the general population. Am J Ophthalmol 50:55, 1960 97. Becker B: Diabetes mellitus and primary open-angle glaucoma. Am J Ophthalmol 71:1, 1971 98. Morgan RW, Drance SM: Chronic open-angle glaucoma and ocular hypertension an epidemiological
study. Br J Ophthalmol 59:211, 1975 99. Katz J, Sommer A: Risk factors for primary open-angle glaucoma. Am J Prev Med 4:110, 1988 100. Klein BEK, Klein R, Jensen SC: Open-angle glaucoma and older-onset diabetes. The Beaver Dam eye study. Ophthalmology 101:1173, 1994 101. Leighton DA, Phillips CI: Systemic blood pressure in glaucoma. Br J Ophthalmol 52:447, 1972 102. Dielemans I, Vingerling JR, Algra D et al: Primary open-angle glaucoma, intraocular pressure, and systemic blood pressure
in the general elderly population. The Rotterdam Study. Ophthalmology 102:54, 1995 103. Tielsch JM, Katz J, Sommer A et al: Hypertension, perfusion pressure, and primary open-angle glaucoma. A population-based
assessment. Arch Ophthalmol 113:216, 1995 104. Wang JJ, Mitchell P, Smith W: Is there an association between migraine headache and open-angle glaucoma? Findings
from the Blue Mountains Eye Study. Ophthalmology 104:1714, 1997 105. Usui T, Iwata K, Shirakashi M et al: Prevalence of migraine in low-tension glaucoma and primary open-angle glaucoma
in Japanese. Br J Ophthalmol 75:224, 1991 106. Phelps CD, Corbett JJ: Migraine and low-tension glaucoma. A case-control study. Invest Ophthalmol Vis Sci 26:1105, 1985 107. Yablonski ME, Zimmerman TJ, Kass MA et al: Prognostic significance of optic disk cupping in ocular hypertensive patients. Am J Ophthalmol 89:585, 1980 108. Martin MJ, Sommer A, Gold EB et al: Race and primary open-angle glaucoma. Am J Ophthalmol 99:383, 1985 109. Wilson R, Richardson TM, Hertzmark E et al: Race as a risk factor for progressive glaucomatous damage. Ann Ophthalmol 17:653, 1985 110. Drance SM, Fairclough M, Butler DM et al: The importance of disc hemorrhage in the prognosis of chronic open-angle
glaucoma. Arch Ophthalmol 95:226, 1977 111. Shihab ZM, Lee PH, Hay P: The significance of disc hemorrhage in open-angle glaucoma. Ophthalmology 89:211, 1982 112. Katavisto M: The diurnal variations of ocular tension in glaucoma. Acta Ophthalmol Suppl 78:1, 1964 113. Newell FW, Krill AE: Diurnal tonography in normal and glaucomatous eyes. Trans Am Ophthalmol Soc 62:349, 1964 114. Kitazawa Y, Horie T: Diurnal variation of intraocular pressure in primate open-angle glaucoma. Am J Ophthalmol 79:557, 1975 115. David R, Zangwill L, Briscoe D et al: Diurnal intraocular pressure variations: An analysis of 690 diurnal curves. Br J Ophthalmol 76:280, 1992 116. Weitzman ED, Henkind P, Leitman M et al: Correlative 24-hour relationships between intraocular pressure and plasma
cortisol in normal subjects and patients with glaucoma. Br J Ophthalmol 59:566, 1975 117. Sheridan PT, Brubaker RF, Larsson L-I et al: The effect of oral dexamethasone on the circadian rhythm of aqueous humor
flow in humans. Invest Ophthalmol Vis Sci 35: 1150, 1994 118. Jonas JB, Gusek GC, Guggenmoos-Holzmann I et al: Variability of the real dimensions of normal human optic discs. Graefes Arch Clin Exp Ophthalmol 226:332, 1988 119. Jonas JB, Gusek GC, Naumann GOH: Optic disc, cup and neuroretinal rim size, configuration and correlations
in normal eyes. Invest Ophthalmol Vis Sci 29:1151, 1988 120. Schwartz JT, Reuling FH, Garrison RJ: Acquired cupping of the optic nerve head in normotensive eyes. Br J Ophthalmol 59:216, 1975 121. Carpel EF, Engstrom PF: The normal cup-disk ratio. Am J Ophthalmol 91:588, 1981 122. Fishman RS: Optic disc asymmetry. A sign of ocular hypertension. Arch Ophthalmol 84:590, 1970 123. Holm OC, Becker B, Asseff CF et al: Volume of the optic disk cup. Am J Ophthalmol 73:876, 1972 124. Armaly MF: Genetic determination of cup/disc ratio of the optic nerve. Arch Ophthalmol 78:35, 1967 125. Teikari JM, Airaksinen JP: Twin study on cup/disc ratio of the optic nerve head. Br J Ophthalmol 76:218, 1980 126. Nicolela MT, Drance SM: Various glaucomatous optic nerve appearances. Clinical Correlations. Ophthalmology 103:640, 1996 127. Spaeth GL, Hitchings RA, Sivalingam E: The optic disc in glaucoma: Pathogenetic correlation of five patterns of
cupping in chronic open-angle glaucoma. Trans Am Acad Ophthalmol Otol 81:217, 1976 128. Hitchings RA, Spaeth GL: The optic disc in glaucoma. I: classification. Br J Ophthalmol 60:778, 1976 129. Radius RL, Maumenee AE, Green WF: Pit-like changes of the optic nerve head in open-angle glaucoma. Br J Ophthalmol 62:389, 1978 130. Jonas JB, Xu L: Optic disk hemorrhages in glaucoma. Am J Ophthalmol 118:1, 1994 131. Hendricks KH, van den Enden A, Rasker MT et al: Cumulative incidence of patients with disc hemorrhages in glaucoma and
the effect of therapy. Ophthalmology 101:1165, 1994 132. Healey RP, Mitchell P, Smith W et al: Optic disc hemorrhages in a population with and without signs of glaucoma. Ophthalmology 105:216, 1998 133. Tuulonen A, Takamoto T, Wu D-C et al: Optic disc cupping and pallor measurements of patients with disk hemorrhages. Am J Ophthalmol 103:505, 1987 134. Airaksinen PJ, Mustonen E, Alanko HI: Optic disc hemorrhages precede retinal nerve fiber layer defects in ocular
hypertension. Acta Ophthalmol 59:627, 1981 135. Bengtsson B, Holmin C, Krakau CET: Disc hemorrhage and glaucoma. Acta Ophthalmol 59:1, 1981 136. Bengtsson B: Optic disc haemorrhages preceding manifest glaucoma. Acta Ophthalmol 108:545, 1990 137. Quigley HA, Miller NR, George T: Clinical evaluation of nerve fiber layer atrophy as an indicator of glaucomatous
optic nerve damage. Arch Ophthalmol 98:1564, 1984 138. Jonas JB, Schiro D: Localised wedge-shaped defects of the retinal nerve fibre layer in glaucoma. Br J Ophthalmol 78:285, 1994 139. Airaksinen PJ, Tuulonen A: Early glaucoma changes in patients with and without optic disc hemorrhage. Acta Ophthalmol 62:197, 1984 140. Sommer A, Miller NR, Pollack I et al: The nerve fiber layer in the diagnosis of glaucoma. Arch Ophthalmol 95:2149, 1977 141. Sommer A, Pollack I, Maumenee AE: Optic disc parameters and onset of glaucomatous field loss. II. Static
screening criteria. Arch Ophthalmol 97:1449, 1979 142. Quigley HA, Katz J, Derick RJ et al: An evaluation of optic disc and nerve fiber layer examinations in monitoring
progression of early glaucoma damage. Ophthalmology 99:19, 1992 143. Jonas JB, Königsbrether KA: Optic disk appearance in ocular hypertensive eyes. Am J Ophthalmol 117:732, 1994 144. Sommer A, Katz J, Quigley HA et al: Clinically detectable nerve fiber atrophy precedes the onset of glaucomatous
field loss. Arch Ophthalmol 109:77, 1991 145. Jonas JB, Naumann OH: Parapapillary chorioretinal atrophy in normal and glaucoma eyes. II. Correlations. Invest Ophthalmol Vis Sci 30:919, 1989 146. Jonas JB, Fernández MC, Naumann GOH: Parapapillary atrophy and retinal vessel diameter in nonglaucomatous optic
nerve damage. Invest Ophthalmol Vis Sci 32:2942, 1991 147. Rockwood EJ, Anderson DR: Acquired peripapillary changes and progression in glaucoma. Graefes Arch Clin Exp Ophthalmol 226:510, 1988 148. Pagon RA: Ocular coloboma. Surv Ophthalmol 25:223, 1981 149. Apple DJ, Rabb MF, Walsh PM: Congenital anomalies of the optic disc. Surv Ophthalmol 27:3, 1982 150. Brodsky MC: Congenital optic disk anomalies. Surv Ophthalmol 39:51, 1980 151. Brazitikos PD, Safran AB, Simona F et al: Threshold perimetry in tilted disc syndrome. Arch Ophthalmol 108:1698, 1990 152. Quigley HA, Anderson DR: Cupping of the optic disc in ischemic optic neuropathy. Trans Am Acad Ophthalmol Otol 83:755, 1977 153. Sebag J, Thomas JV, Epstein DL et al: Optic disc cupping in arteritic anterior ischemic optic neuropathy resembles
glaucomatous cupping. Ophthalmology 93:357, 1986 154. Portney GL, Roth AM: Optic cupping caused by an intracranial aneurysm. Am J Ophthalmol 84:98, 1977 155. Quigley HA, Addicks EM, Green WR: Optic nerve damage in human glaucoma. III. Quantitative correlation of
nerve fiber loss and visual field defect in glaucoma, ischemic neuropathy, papilledema, and
toxic neuropathy. Arch Ophthalmol 100:135, 1982 156. Quigley HA, Dunkelberger GR, Green WR: Retinal ganglion cell atrophy correlated with automated perimetry in human
eyes with glaucoma. Am J Ophthalmol 107:453, 1989 157. Johnson CA, Adams AJ, Casson EJ et al: Blue-on-yellow perimetry can predict the development of glaucomatous visual
field loss. Arch Ophthalmol 111:645, 1993 158. Johnson CA, Brandt JD, Khong AM et al: Short-wavelength automated perimetry in low-, medium-, and high-risk ocular
hypertensive eyes. Arch Ophthalmol 113:70, 1995 159. Wall M, Jennisch CS, Munden PM: Motion perimetry identifies nerve fiber bundle-like defects in ocular hypertension. Arch Ophthalmol 115:26, 1997 160. Korth M, Hor F, Storck B et al: Spatial and spatiotemporal contrast sensitivity of normal and glaucoma
eyes. Graefes Arch Clin Exp Ophthalmol 227:428, 1989 161. Wanger P, Persson HE: Pattern-reversal electroretinograms and high-pass resolution perimetry
in suspected or early glaucoma. Ophthalmology 94:1098, 1987 162. Weinstein GW, Arden GB, Hitchings RA et al: The pattern electroretinogram (PERG) in ocular hypertension and glaucoma. Arch Ophthalmol 106:923, 1988 163. Weinreb RN: Diagnosing and monitoring glaucoma with confocal scanning laser ophthalmoscopy. J Glaucoma 4: 225, 1995 164. Anton A, Zangwill L, Embadi A et al: Nerve fiber layer measurements with scanning laser polarimetry in ocular
hypertension. Arch Ophthalmol 115:331, 1997 165. Weinreb RN, Shakiba S, Zangwill L: Scanning laser polarimetry to measure the nerve fiber layer of normal and
glaucomatous eyes. Am J Ophthalmol 119:627, 1995 166. Schuman JS, Hee MR, Puliafito CA et al: Quantification of nerve fiber layer-thickness in normal and glaucomatous
eyes using optical coherence tomography. Arch Ophthalmol 113:586, 1995 167. Caprioli J: Recognizing structural damage to the optic nerve head and nerve fiber layer
in glaucoma. Am J Ophthalmol 124:516, 1997 168. Jay JL, Allan D: The benefit of early trabeculectomy versus conventional management in primary
open-angle glaucoma relative to severity of disease. Eye 3:528, 1989 169. Migdal C, Gregory W, Hitchings R: Long-term functional outcome after early surgery compared with laser and
medicine in open-angle glaucoma. Ophthalmology 101:1651, 1994 170. Odberg T: Visual field prognosis in advanced glaucoma. Acta Ophthalmol 65(suppl 182):27, 1987 171. Mao LK, Steward WC, Shields MB: Correlation between intraocular pressure control and progressive glaucomatous
damage in primary open-angle glaucoma. Am J Ophthalmol 111:51, 1991 172. Kolker AE: Visual prognosis in advanced glaucoma: A comparison of medical and surgical
therapy for retention of vision in 101 eyes with advanced glaucoma. Trans Am Ophthalmol Soc 75:539, 1977 173. Quigley HA, Maumenee AE: Long-term follow-up of treated open-angle glaucoma. Am J Ophthalmol 87:519, 1979 174. Glaucoma Laser Trial Research Group: The Glaucoma Laser Trial (GLT) and
Glaucoma Laser Trial Follow-up Study: 7. Results. Am J Ophthalmol 120:718, 1996 175. Wilson MR, Gaasterland D: Translating research into practice: Controlled clinical trials and their
influence on glaucoma management. J Glaucoma 5(2):139, 1996 176. Coakes RL, Brubaker RF: The mechanism of timolol in lowering intraocular pressure in the normal
eye. Arch Ophthalmol 96:2045, 1978 177. Bartels SP, Roth O, Jumblatt MM et al: Pharmacological effects of topical timolol in the rabbit eye. Invest Ophthalmol Vis Sci 19:1189, 1980 178. Maclure GM: Chronic open angle glaucoma treated with timolol. A four-year study. Trans Ophthalmol Soc UK 103:78, 1983 179. Boger WP III, Puliafito CA, Steinert RF et al: Long-term experience with timolol ophthalmic solution in patients with
open-angle glaucoma. Ophthalmology 85:259, 1978 180. Van Buskirk EM, Fraunfelder FT: Ocular beta-blockers and systemic effects. Am J Ophthalmol 98:623, 1984 181. Allen RC, Hertzmark E, Walker AM et al: A double-masked comparison of betaxolol vs timolol in the treatment of
open-angle glaucoma. Am J Ophthalmol 101:535, 1986 182. Schoene RB, Abuan T, Ward RL et al: Effects of topical betaxolol, timolol, and placebo on pulmonary function
in asthmatic bronchitis. Am J Ophthalmol 97:86, 1984 183. Sears ML: Autonomic nervous system: adrenergic agonists. In Sears ML (ed): Handbook
of Experimental Pharmacology, vol 69. Berlin, Springer-Verlag, 1984 184. Alexander DW, Berson FG, Epstein DL: A clinical trial of timolol and epinephrine in the treatment of primary
open-angle glaucoma. Ophthalmology 95:247, 1988 185. Sonntag JR, Brindley GO, Shields MB et al: Timolol and epinephrine. Comparison of efficacy and side effects. Arch Ophthalmol 97:273, 1979 186. Veirs ER, McGrew JC: Ocular complications from topical epinephrine therapy of glaucoma. EENT Monthly 42:46, 1963 187. Wei C-P, Anderson JA, Leopold I: Ocular absorption and metabolism of topically applied epinephrine and dipivalyl
ester of epinephrine. Invest Ophthalmol Vis Sci 17:315, 1978 188. Kerr CR, Hass I, Drance Sm et al: Cardiovascular effects of epinephrine and dipivalyl epinephrine applied
topically to the eye in patients with glaucoma. Br J Ophthalmol 66:109, 1982 189. Toris CB, Tafoya ME, Camras CB et al: Effects of apraclonidine on aqueous humor dynamics in human eyes. Ophthalmology 102:456, 1995 190. Toris CB, Gleason ML, Camras CB et al: Effects of brimonidine on aqueous humor dynamics in human eyes. Arch Ophthalmol 113:1514, 1995 191. Nagasubramanian S, Hitchings RA, Demailly P et al: Comparison of apraclonidine and timolol in chronic open-angle glaucoma. A
three-month study. Ophthalmology 100: 1318, 1993 192. Schuman JS: Clinical experience with brimonidine 0.2% and timolol 0.5% in glaucoma
and ocular hypertension. Surv Ophthalmol 41:S27, 1996 193. Robin AL: Short-term effects of unilateral 1% apraclonidine therapy. Arch Ophthalmol 106:912, 1988 194. Butler P, Mannschreck M, Lin S et al: Clinical experience with the long-term use of 1% apraclonidine. Arch Ophthalmol 113:293, 1995 195. Kaufman PL, Bárány EH: Loss of acute pilocarpine effect on outflow facility following surgical
disinsertion and retrodisplacement of the ciliary muscle from the scleral
spur in the cynomolgus monkey. Invest Ophthalmol 15:793, 1976 196. Bill A, Phillips CI: Uveoscleral drainage of aqueous humour in human eyes. Exp Eye Res 12:275, 1971 197. Boger WP III, Steinert RF, Puliafito CA et al: Clinical trial comparing timolol ophthalmic solution to pilocarpine in
open-angle glaucoma. Am J Ophthalmol 86:8, 1978 198. Christakis C, Mangouritsas N: Comparative studies of the pressure-lowering effect of timolol and phospholine
iodide. Klin Monastsbl Augenheilkd 179:197, 1981 199. Ellis PP, Esterdahl M: Echothiophate iodide therapy in children. Effect upon blood cholinesterase
levels. Arch Ophthalmol 77:598, 1967 200. Constant MA, Becker B: The effect of carbonic anhydrase inhibitors on urinary excretion of citrate
by humans. Am J Ophthalmol 49:929, 1960 201. Bietti G, Virno M, Pecori-Giraldi J et al: Acetazolamide, metabolic acidosis, and intraocular pressure. Am J Ophthalmol 80:360, 1975 202. Dailey RA, Brubaker RF, Bourne WM: The effects of timolol maleate and acetazolamide on the rate of aqueous
formation in normal human subjects. Am J Ophthalmol 93:232, 1982 203. Lichter PR: Reducing side effects of carbonic anhydrase inhibitors. Ophthalmology 88:266, 1981 204. Strahlman E, Tipping R, Vogel R et al: A double-masked, randomized 1-year study comparing dorzolamide (Trusopt), timolol, and
betaxolol. Arch Ophthalmol 113:1009, 1995 205. Luetjen-Drecoll E, Tamm E: Morphological study of the anterior segment of cynomolgus monkey eyes following
treatment with prostaglandin F2a. Exp Eye Res 47:761, 1988 206. Camras CB: Comparison of latanoprost and timolol in patients with ocular hypertension
and glaucoma. A six-month masked, multicenter trial in the United
States. Ophthalmology 103:138, 1996 207. Alm A, Stjernschantz J. Scandinavian Latanoprost Study Group: Effect on
intraocular pressure and side effects of 0.005% latanoprost applied once
daily, evening or morning. A comparison with timolol. Ophthalmology 102:1743, 1995 208. Warwar RE, Bullock JD, Ballal D: Cystoid macular edema and anterior uveitis associated with latanoprost
use: Experience and incidence in a retrospective review of 94 patients. Ophthalmology 105:263, 1998 209. Kolker AE: Hyperosmotic agents in glaucoma. Invest Ophthalmol 9:418, 1970 210. Wise JB: Long-term control of adult open angle glaucoma by argon laser treatment. Ophthalmology 88:197, 1981 211. Wise JB, Witter SL: Argon laser therapy for open-angle glaucoma. A pilot study. Arch Ophthalmol 97:319, 1979 212. Alexander RA, Grierson I, Church WH: The effect of argon laser trabeculoplasty upon the normal human trabecular
meshwork. Graefes Arch Clin Exp Ophthalmol 227:72, 1989 213. Eendebak GR, Boen-Tan TN, Bezemer PD: Long-term follow-up of laser trabeculoplasty. Doc Ophthalmol 75: 203, 1990 214. Wise JB: Ten-year results of laser trabeculoplasty. Eye 1: 45, 1987 215. Forbes M, Bansal RK. Argon laser goniophotocoagulation of the trabecular
meshwork in open angle glaucoma. Trans Am Ophthalmol Soc 79:257, 1981 216. Thomas JV, Simmons RJ, Belcher CD: Argon laser trabeculoplasty in the pre-surgical glaucoma patient. Ophthalmology 89:187, 1982 217. Levene R: Major early complications of laser trabeculoplasty Ophthalmic
Surg 14:947, 1983 218. Weinreb RN, Ruderman J, Juster R et al: Immediate intraocular pressure response to argon laser trabeculoplasty. Am J Ophthalmol 95:279, 1983 219. Holmwood PC, Chase RD, Krupin T et al: Apraclonidine and argon laser trabeculoplasty. Am J Ophthalmology 114:19, 1992 220. Barnebey HS, Robin AL, Zimmerman TJ et al: The efficacy of brimonidine in decreasing elevation in intraocular pressure
after laser trabeculoplasty. Ophthalmology 100:1083, 1993 221. Hovanesian JAD, Higginbotham E, Lichter P et al: Long-term visual outcome of ocular hypotension after thermosclerostomy. Am J Ophthalmol 115:603, 1993 222. Lewis RA, Phelps CD: Trabeculectomy v. thermosclerostomy—A five year follow-up. Arch Ophthalmol 102:533, 1984 223. Marion JR, Shields MB: Thermal sclerostomy and posterior lip sclerectomy: a comparative study. Ophthalmic Surg 9: 67, 1978 224. Benscsik R, Opauzki A, Hudomel I: Effectiveness of trepanotrabeculectomy in glaucoma. Glaucoma 3:42, 1981 225. Cairns JE: Trabeculectomy—preliminary report of a new method. Am J Ophthalmol 66:673, 1968 226. Nouri-Mahdavi K, Brigatti L, Weitzman M et al: Outcomes of trabeculectomy for primary open-angle glaucoma. Ophthalmology 102:1760, 1995 227. Fluorouracil Filtering Surgery Study Group T: Three-year follow-up of the
fluorouracil filtering surgery study. Am J Ophthalmol 115:82, 1993 228. Skuta GL, Beeson CC, Higginbotham EJ: Intraoperative mitomycin versus postoperative 5-fluorouracil in high-risk
glaucoma filtering surgery. Ophthalmology 99:438, 1992 229. Zacharia PT, Deppermann SR, Schuman JS: Ocular hypotony after trabeculectomy with mitomycin C. Am J Ophthalmol 116:314, 1993 230. Ticho U, Ophir A: Late complications after glaucoma filtering surgery. Am J Ophthalmol 115:506, 1993 231. Stamper RL, McMenemy MG, Lieberman MF: Hypotonous maculopathy after trabeculectomy with subconjunctival 5-fluorouracil. Am J Ophthalmol 114:544, 1992 232. Lee DA, Hersh P, Kersten D et al: Complications of subconjunctival 5-fluorouracil following glaucoma filtration
surgery. Ophthalmic Surg 18:187, 1987 233. Downes RN, Flanagan DW, Jordan K et al: The Molteno implant in intractable glaucoma. Eye 2:250, 1988 234. Heuer DK, Lloyd MA, Abrams DA et al: Which is better? One or Two? A randomized clinical trial of single-plate
versus double-plate Molteno implantation for glaucomas in aphakia and
pseudophakia. Ophthalmology 99:1512, 1992 235. Hodkin MJ, Goldblatt WS, Burgoyne CF et al: Early clinical experience with the Baerveldt implant in complicated glaucoma. Am J Ophthalmol 120:32, 1995 236. Krupin Eye Valve Filtering Surgery Study Group: Krupin eye valve with disc
for filtration surgery. Ophthalmology 101:651, 1994 237. Coleman AL, Hill R, Wilson MR et al: Initial clinical experience with the Ahmed Glaucoma Valve implant. Am J Ophthalmol 120:23, 1995 238. Melamed S, Cahane M, Gutman I et al: Postoperative complications after Molteno implant surgery. Am J Ophthalmol 111:319, 1991 239. Quigley HA: Histological and physiological studies of cyclocryotherapy in primate and
human eyes. Am J Ophthalmol 82:722, 1976 240. Caprioli J, Strang SL, Spaeth GL et al: Cyclocryotherapy in the treatment of advanced glaucoma. Ophthalmology 92:947, 1985 241. Brindley G, Shields MB: Value and limitations of cyclocryotherapy. Graefes Arch Clin Exp Ophthalmol 224:545, 1986 242. Balazsi G: Noncontact thermal mode Nd:YAG laser transscleral cyclocoagulation in the
treatment of glaucoma. Ophthalmology 98:1858, 1991 243. Trope GE, Ma S: Mid-term effects of neodymium:YAG transscleral cyclocoagulation in glaucoma. Ophthalmology 97:73, 1990 244. Brancato R, Giovanni L, Trabucchi G et al: Contact transscleral cyclophotocoagulation with Nd:YAG laser in uncontrolled
glaucoma. Ophthalmic Surg 20:547, 1989 245. Kosoko O, Gaasterland DE, Pollack IP et al: Long-term outcome of initial ciliary ablation with contact diode laser
transscleral cyclophotocoagulation for severe glaucoma. Ophthalmology 103:1294, 1996 246. Shields S, Stewart WC, Shields MB: Transpupillary argon laser cyclophotocoagulation in the treatment of glaucoma. Ophthalmic Surg 19:171, 1988 247. Uram M: Ophthalmic laser microendoscope endophotocoagulation. Ophthalmology 99:1829, 1992 248. Yamishita H, Sears ML: Complications of cyclocryosurgery. Glaucoma 2:273, 1980 249. Maus M, Katz LJ: Choroidal detachment, flat anterior chamber, and hypotony as complications
of neodymium. YAG laser cyclophotocoagulation. Ophthalmology 97:69, 1990 250. Flammer J, Guthauser U, Mahler F: Do ocular vasospasms help cause low tension
glaucoma? In Greve EL, Heijl A (eds): Seventh International Visual
Field Symposium, Amsterdam, September 1986, Martinus Nijhoff/Dr W. Junk
Publishers, Dordrecht, Netherlands, 1987 251. Rojanapongpun P, Drance SM: The response of blood flow velocity in the ophthalmic artery and blood
flow of the finger to warm and cold stimuli in glaucomatous patients. Graefes Arch Clin Exp Ophthalmol 231(7):375, 1993 252. Netland PA, Chaturvedi N, Dreyer EB: Calcium channel blockers in the management of low-tension and open-angle
glaucoma. Am J Ophthalmol 115:608, 1993 253. Dreyer EB, Lipton SA: Excitatory amino acids in glaucoma: A potentially novel etiology of neuronal
loss. Invest Ophthalmol Vis Sci 33:1093, 1992 254. Schumer RA, Podos SM, Lipton SA et al: Increased glutamate in the vitreous of monkeys with induced glaucoma. Invest Ophthalmol Vis Sci 35:1484, 1994 255. Caprioli J, Morgan J, Kitano S: Hyperthermia and hypoxia increase tolerance of retinal ganglion cells to
anoxia and excitotoxicity. Invest Ophthalmol Vis Sci 37:2376, 1996 256. Moncada S, Palmer R, Higgs EA: Nitric oxide physiology, pathophysiology, and pharmacology. Pharmacol Rev 43: 109, 1991 257. Pellegrini-Giampietro D, Cherici G, Alesiani M et al: Excitatory amino acid release and free radical formation may cooperate
in the genesis of ischemia-induced neuronal damage. J Neurosci 10:1035, 1990 258. McNamara J, Fridovich I: Did radicals strike Lou Gehrig? Nature 362:20, 1993 259. Lam TT, Fu J, Hrynewycz M et al: The effect of aurintricarboxylic acid, an endonuclease inhibitor, on ischemia/ reperfusion
damage in rat retina. J Ocul Pharmacol Ther 11:253, 1995 260. Villegas-Perez MP, Vidal-Sanz M, Bray GM et al: Influences of peripheral grafts on the survival and regrowth of axotomized
retinal ganglion cells in adult rats. J Neurosci 8:265, 1988 261. Aguayo AJ, Bray GM, Rasminsky M et al: Synaptic connections made by axons regenerating in the central nervous
system of adult mammals. J Exp Biol 153:199, 1990 262. Jampel HD: Laser trabeculoplasty is the treatment of choice for chronic open-angle
glaucoma. Arch Ophthalmol 116: 240, 1998 263. Hitchings R: Surgery is the treatment of choice for open-angle glaucoma. Arch Ophthalmol 116:241, 1998 264. Schwartz AL: Argon laser trabeculoplasty in glaucoma: What's happening (survey
results of American Glaucoma Society members). J Glaucoma 2:329, 1993 265. Higginbotham EJ: Medication is the treatment of choice for chronic open-angle glaucoma. Arch Ophthalmol 116: 239, 1998 |  50
50