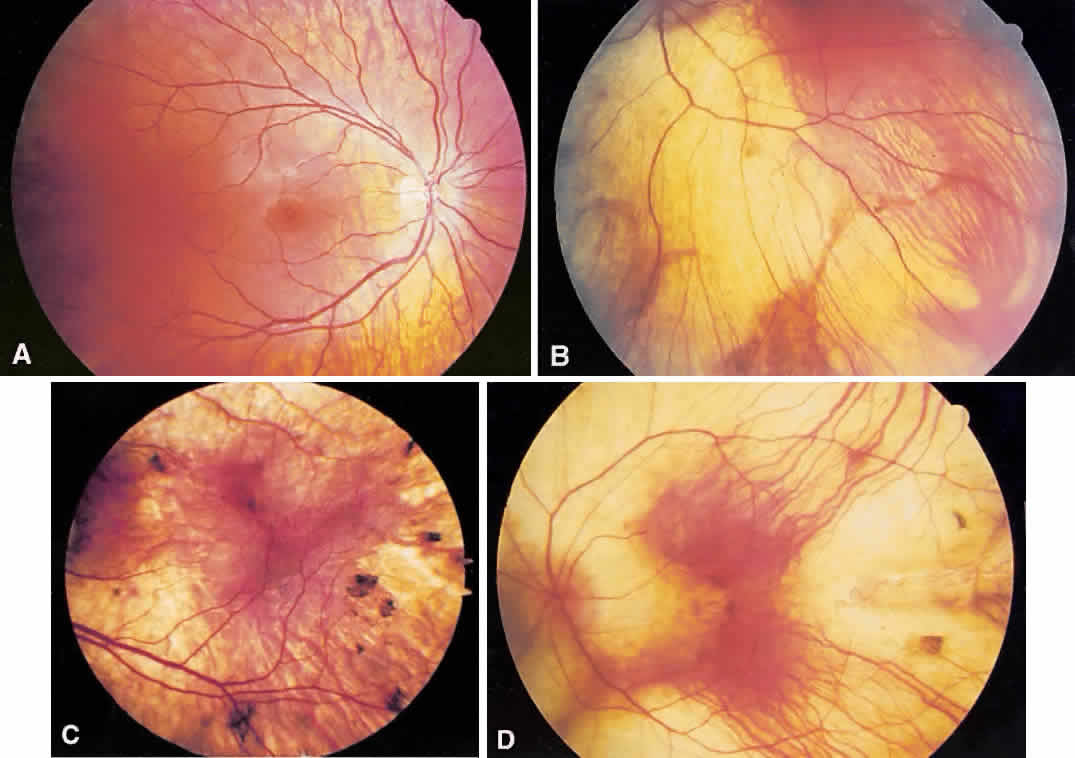
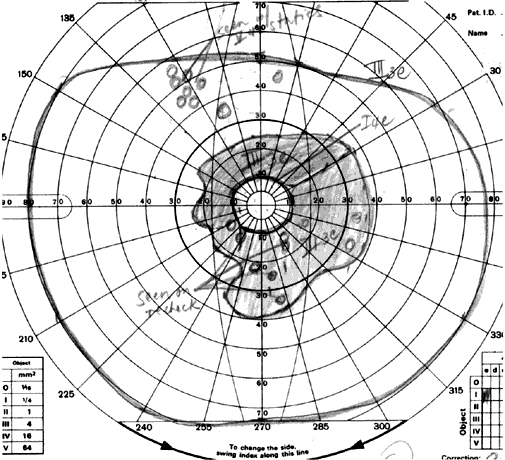
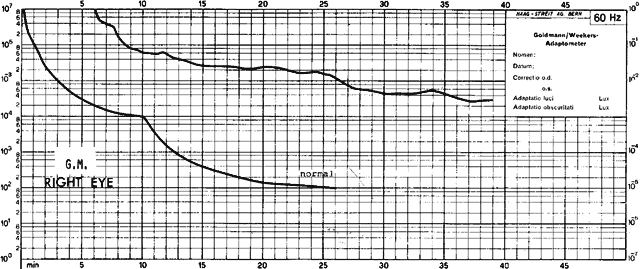
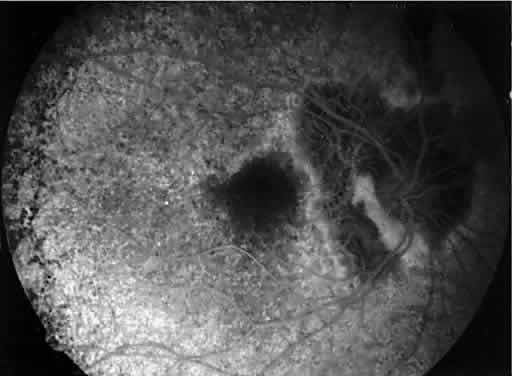
1. Mauthner H: Ein Fall von Choroideremi. Berl Nat Med Innsbruck 2:191, 1872 2. McCulloch C, McCulloch R: A hereditary and clinical study of choroideremia. Trans Am Acad Ophthalmol Otolaryngol 52:160, 1948 3. Pameyer J, Waardenburg P, Henkes H: Choroideremia. Br J Ophthalmol 44:724, 1960 4. van den Hurk JA, Schwartz M, van Bokhoven H et al: Molecular basis of choroideremia (CHM): Mutations involving the Rab escort
protein-1 (REP-1) gene. Hum Mutat 9:110, 1997 5. van den Hurk JA, Hendriks W, van de Pol DJ et al: Mouse choroideremia gene mutation causes photoreceptor cell degeneration
and is not transmitted through the female germline. Hum Mol Genet 6:851, 1997 6. McDonald I: Choroideremia. New York: Oxford University Press, 1998 7. Abramson D, Fishman H: Chromosomal studies in choroi-deremia. J Pediatr Ophthalmol Strabismus 10:178, 1973 8. Gordon D: Choroideremia. Med Radiogr Photogr 28:110, 1952 9. Karna J: Choroideremia: A clinical and genetic study of 84 Finnish patients and 126 female
carriers. Acta Ophthalmol Suppl 176:1, 1986 10. Heckenlively J: The frequency of posterior subcapsular cataract in the hereditary retinal
degenerations. Am J Ophthalmol 93:733, 1982 11. Cherkunov B, Panormova N: Progressive chorioretinal dystrophy (choroideremia) traced down in three
generations. Vestn Oftalmol 1:70, 1967 12. Krill A: Krill's Hereditary Retinal and Choroidal Diseases. Hagerstown, MD: Harper & Row, 1977 13. Heckenlively J: Choroideremia. In Heckenlively JR, Arden GB, (eds): Principles
and Practice of Clinical Electrophysiology of Vision. St Louis: Mosby—Year
Book, 1991 14. Sieving PA, Niffenegger JH, Berson EL: Electroretinographic findings in selected pedigrees with choroideremia. Am J Ophthalmol 101:361, 1986 15. Kurstjens J: Choroideremia and gyrate atrophy of the choroids and retina. Doc Ophthalmol 19:1, 1965 16. Krill A, Archer D: Classification of the choroidal dystrophies. Am J Ophthalmol 72:187, 1971 17. Forsius H, Hyvarinen L, Nieminen H et al: Fluorescein and indocyanine green fluorescence angiography in the study
of affected males and in female carriers with choroideremia. Acta Ophthalmol 55:459, 1977 18. Noble KG, Carr RE, Siegel IM: Fluorescein angiography of the hereditary choroidal dystrophies. Br J Ophthalmol 61:43, 1977 19. Gass J: Stereoscopic Atlas of Macular Disease: Diagnosis and Treatment. St
Louis: Mosby, 1997 20. Ohba N: Choroideremia: Study of two Japanese families. Acta Soc Ophthalmol Japan 78:116, 1974 21. Cameron J, Fine B, Shapiro I: Histopathologic observations in choroideremia with emphasis on vascular
changes of the uveal tract. Ophthalmology 94:187, 1987 22. Rodrigues M, Ballintine E, Wiggert B et al: Choroideremia: A clinical, electron microscopic and biochemical report. Ophthalmology 91:873, 1984 23. Goedbloed J: Mode of inheritance of choroideremia. Ophthalmologica 104:308, 1942 24. Waardenburg P: Choroideremia als Erbuerknal (zur atrophic). Acta Ophthalmol 20:235, 1942 25. Lyon M: Sex chromatin and gene action in the mammalian X-chromosome. Am J Hum Genet 14:135, 1962 26. Lewis RA, Nussbaum RL, Ferrell R: Mapping X-linked ophthalmic diseases: Provisional assignment of the locus
for choroideremia to Xq13-q24. Ophthalmology 92:800, 1985 27. Cremers F, van de Pol O, van Kerkhoff L et al: Cloning of a gene that is rearranged in patients with choroideremia. Nature 347:674, 1990 28. Merry DE, Janne PA, Landers JE et al: Isolation of a candidate gene for choroideremia. Proc Natl Acad Sci USA 89:2135, 1992 29. Seabra MC, Brown MS, Goldstein JL: Retinal degeneration in choroideremia: deficiency of rab geranylgeranyl
transferase. Science 259:377, 1993 30. Tolmachova T, Ramalho JS, Anant JS et al: Cloning, mapping and characterization of the human RAB27A gene. Gene 239:109, 1999 31. Seabra MC, Ho YK, Anant JS: Deficient geranylgeranylation of Ram/Rab27 in choroideremia. J Biol Chem 270:24420, 1995 32. Cremers F, Molloy C, van de Pol D et al: An autosomal homologue of the choroideremia gene colocalizes with the Usher
syndrome type II locus on the distal part of chromosome 1q. Hum Mol Genet 1:71, 1992 33. Cremers FP, Armstrong SA, Seabra MC et al: REP-2, a Rab escort protein encoded by the choroideremia-like gene.J Biol Chem 269:2111, 1994 34. Fraser B, Friedmann A: Choroideremia in a female. BMJ 2:732, 1968 35. Harris G, Miller J: Choroideremia: Visual defects in a heterozygote. Arch Ophthalmol 80:423, 1968 36. Burke M, Choromokos E, Bibler L et al: Choroideremia in a genotypically normal female: A case report. Ophthalmic Pediatr Genet 6:163, 1985 37. Francois J: Importance of electrophysiology in ophthalmogenetics. Ophthalmologica 188:14, 1984 38. Cheng C, Chen M, Hou P: Choroideremia: A study of two families. J Formos Med Assoc 90:1103, 1991 39. Pinckers A, van Aarem A, Brink H: The electrooculogram in heterozygote carriers of Usher syndrome, retinitis
pigmentosa, neuronal ceroid lipofuscinosis, senior syndrome and choroideremia. Ophthalmic Genet 15:25, 1994 40. MacDonald I, Chen M, Addison D, Mielke B et al: Histo-pathology of the retinal pigment epithelium of a female carrier of
choroideremia. Can J Ophthalmol 32:329, 1997 41. Flannery J, Bird A, Farber D et al: A histopathologic study of a choroideremia carrier. Invest Ophthalmol Vis Sci 31:229, 1990 42. Ghosh M, McCullock C, Parker J: Pathological study ina female carrier of choroideremia. Can J Ophthalmol 23:181, 1988 43. MacDonald IM, Mah DY, Ho YK et al: A practical diagnostic test for choroideremia. Ophthalmology 105:1637, 1998 44. Beaufrere L, Tuffery S, Hamel C et al: The protein truncation test (PTT) as a method of detection for choroideremia
mutations. Exp Eye Res 65:849, 1997 45. Hayakawa M, Fujiki K, Hotta Y et al: Visual impairment and REP-1 gene mutations in Japanese choroideremia patients. Ophthalmic Genet 20:107, 1999 46. van Bokhoven H, Schwartz M, Andreasson S et al: Mutation spectrum in the CHM gene of Danish and Swedish choroide-remia
patients. Hum Mol Genet 3:1047, 1994 47. Beaufrere L, Tuffery S, Hamel C et al: A novel mutation (S558X) causing choroideremia. Hum Mutat 8:395, 1996 48. Beaufrere L, Tuffery S, Hamel C et al: Rapid genetic diagnosis of females carriers related to patients with choroideremia. (original
in French). J Fr Ophtalmol 20:534, 1997 49. Beaufrere L, Tuffery S, Hamel C et al: An exonic polymorphism (381A/G) in the choroideremia gene. Genet Couns 8:223, 1997 50. Beaufrere L, Rieu S, Hache J et al: Altered rep-1 expression due to substitution at position + 3 of the
IVS13 splice choroi-deremia (CHM) gene. Curr Eye Res 17:726, 1998 51. Beaufrere L, Rieu S, Hache JC et al: Length variations of the poly(T) tract at the exon 3 splice acceptor site
of the choroideremia gene. Genet Couns 9:255, 1998 52. Beaufrere L, Girardet A, Arnaud B et al: Update on a diagnostic test for choroideremia: The protein truncation test (PTT) (original
in French). J Fr Ophtalmol 21:345, 1998 53. Beaufrere L, Rieu S, Hache JC et al: Altered rep-1 expression due to substitution at position + 3 of the
IVS13 splice-donor site of the choroideremia (CHM) gene. Curr Eye Res 17:726, 1998 54. Beaufrere L, Claustres M, Tuffery S: No missense mutation in choroideremia patients analyzed to date. Ophthalmic Genet 20:89, 1999 55. Forsythe P, Maguire A, Fujita R et al: A carboxy-terminal truncation of 99 amino
acids resulting from a novel mutation (Arg555 → stop) in
the CHM gene leads to choroideremia. Exp Eye Res 64:487, 1997 (Letter) 56. Fujiki K, Hotta Y, Hayakawa M et al: REP-1 gene mutations in Japanese patients with choroideremia. Graefs Arch Clin Exp Ophthalmol 237:735, 1999 57. Pascal O, Donnelly P, Fouanon C et al: A new (old) deletion in the choroideremia gene. Hum Mol Genet 2:1489, 1993 58. Schwartz M, Rosenberg T, van den Hurk JA et al: Identification of mutations in Danish choroideremia families. Hum Mutat 2:43, 1993 59. Jiminez-Sierra J, Ogden T: Inherited Retinal Diseases: A Diagnostic Guide. St
Louis: CV Mosby, 1989 60. Mitchell GA, Looney JE, Brody LC et al: Human ornithine-delta-aminotransferase: cDNA cloning and analysis of the
structural gene. J Biol Chem 263:14288, 1988 61. McInnes RR, Arshinoff SA, Bell L et al: Hyperornithinaemia and gyrate atrophy of the retina: Improvement of vision
during treatment with a low-arginine diet. Lancet 1:513, 1981 62. Schwahn U, Lenzner S, Dong J et al: Positional cloning of the gene for X-linked retinitis pigmentosa 2. Nat Genet 19:327, 1998 63. Meindl A, Dry K, Herrmann K et al: A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor
is mutated in X-linked retinitis pigmentosa (RP3). Nat Genet 13:35, 1996 64. Ott J, Bhattacharya S, Chen JD et al: Localizing multiple X chromosome-linked retinitis pigmentosa loci using
multilocus homogeneity tests. Proc Natl Acad Sci USA 87:701, 1990 65. Mears AJ, Hiriyanna S, Vervoort R et al: Remapping of the RP15 locus for X-linked cone-rod degeneration to Xp11.4-p21.1, and
identification of a de novo insertion in the RPGR exon ORF15. Am J Hum Genet 67:1000, 2000 66. Hardcastle AJ, Thiselton DL, Zito I et al: Evidence for a new locus for X-linked retinitis pigmentosa (RP23). Invest Ophthalmol Vis Sci 41:2080, 2000 67. Gieser L, Fujita R, Goring HH et al: A novel locus (RP24) for X-linked retinitis pigmentosa maps to Xq26-27. Am J Hum Genet 63:1439, 1998 68. Young TL, Ronan SM, Alvear AB et al: A second locus for familial high myopia maps to chromosome 12q. Am J Hum Genet 63:1419, 1998 69. Young TL, Ronan SM, Drahozal LA et al: Evidence that a locus for familial high myopia maps to chromosome 18p. Am J Hum Genet 63:109, 1998 70. Schwartz M, Haim M, Skarsholm D: X-linked myopia: Bornholm eye disease: Linkage to DNA markers on the distal
part of Xq. Clin Genet 38:281, 1990 71. Bassi MT, Schiaffino MV, Renieri A et al: Cloning of the gene for ocular albinism type 1 from the distal short arm
of the X chromosome. Nat Genet 10:13, 1995 72. Schiaffino MV, d'Addio M, Alloni A et al: Ocular albinism: Evidence for a defect in an intracellular signal transduction
system. Nat Genet 23:108, 1999 73. Ayazi S: Choroideremia, obesity, and congenital deafness. Am J Ophthal 92:63, 1981 74. Menon RK, Ball WS, Sperling MA: Choroideremia and hypopituitarism: An association. Am J Med Genet 34:511, 1989 75. May M, Colleaux L, Murgia A et al: Molecular analysis of four males with mental retardation and deletions
of Xq21 places the putative MR region in Xq21.1 between DXS233 and CHM. Hum Mol Genet 4:1465, 1995 76. Nussbaum RL, Lesko JG, Lewis RA et al: Isolation of anonymous DNA sequences from within a submicroscopic X chromosomal
deletion in a patient with choroideremia, deafness, and mental
retardation. Proc Natl Acad Sci USA 84:6521, 1987 77. Rosenberg T, Niebuhr E, Yang HM et al: Choroideremia, congenital deafness and mental retardation in a family with
an X chromosomal deletion. Ophthalmic Paediatr Genet 8:139, 1987 |