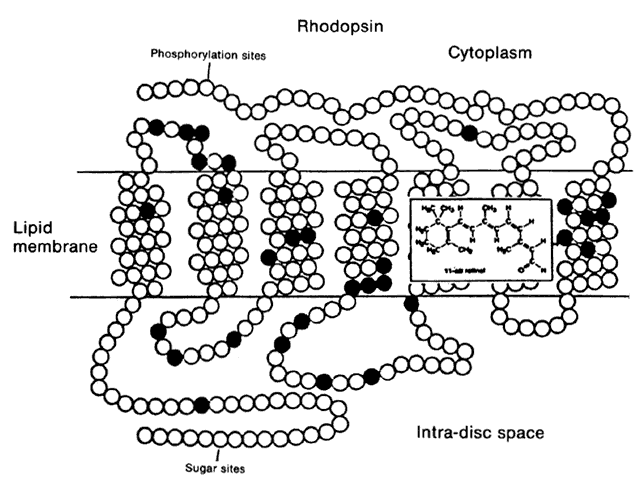
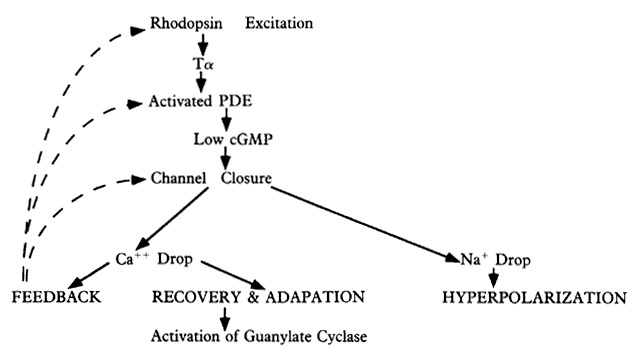
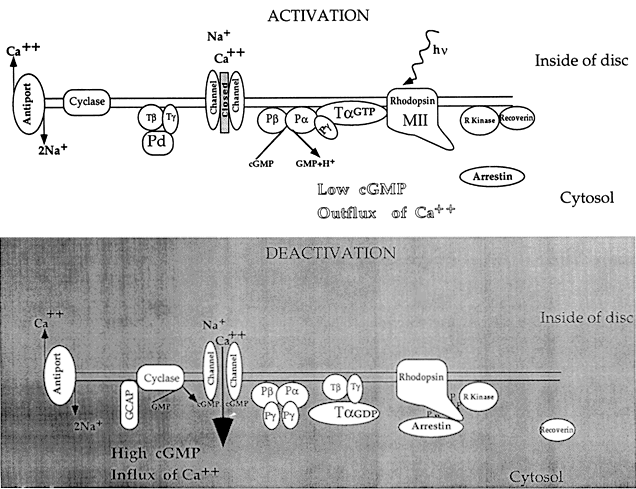
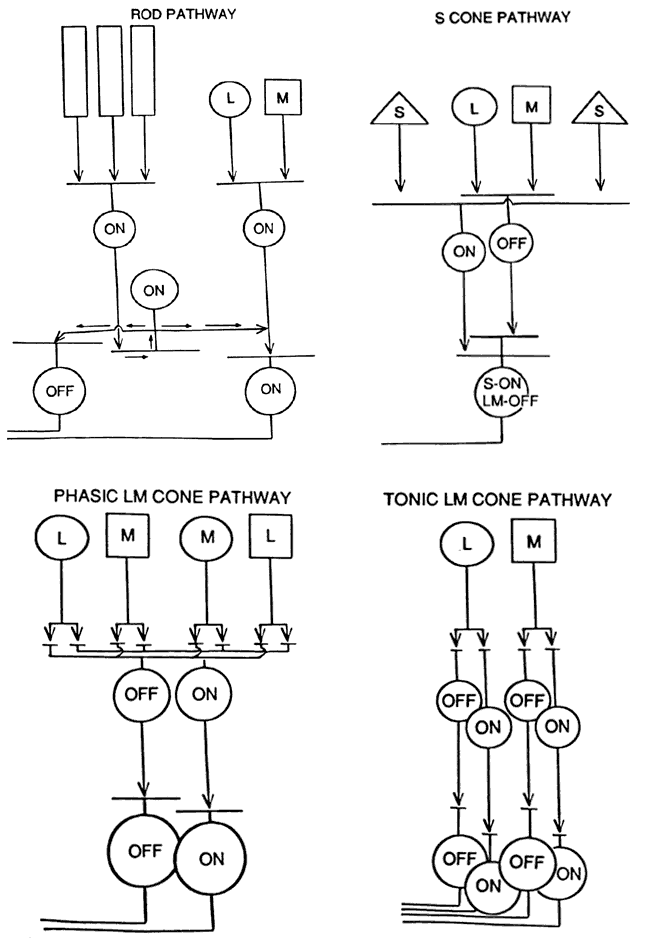
1. Stryer L: Visual excitation and recovery. J Biol Chem 266:10711, 1991 2. Yarfitz S, Hurley JB: Transduction mechanisms of vertebrate and invertebrate photoreceptors. J Biol Chem 269:14329, 1994 3. Fung BK-K, Hurley JB, Stryer L: Flow of information in the light-triggered cyclic nucleotide cascade of
vision. Proc Natl Acad Sci USA 78:152, 1981 4. Baehr W, Devlin MJ, Applebury ML: Isolation of bovine ROS phosphodiesterase. J Biol Chem 254:11669, 1979 5. Fung BKK, Young JH, Yamane HK, Griswold-Prenner I: Subunit stoichiometry of retinal rod cGMP phosphodiesterase. Biochemistry 29:2657, 1990 6. Wensel TG, Stryer L: Reciprocal control of retinal rod cyclic GMP phosphodiesterase by its <gt> subunit
and transducin. Proteins Struct Funct Genet 1:90, 1986 7. Deterre P, Bigay J, Forquet F et al: cGMP phosphodiesterase of retinal rods is regulated by two inhibitory subunits. Proc Natl Acad Sci USA 85:2424, 1988 8. Clerc A, Bennett N: Activation of cGMP phosphodiesterase of retinal rods: A complex with transducin
alpha subunit. J Biol Chem 267:6620, 1992 9. Sitaramayya A, Harkness J, Parkes JH et al: Kinetic studies suggest that light-activated cyclic GMP phosphodiesterase
is a complex with G-protein subunits. Biochemistry 25:651, 1986 10. Arshavsky VY, Bownds DM: Regulation of deactivation of photoreceptor G protein by its target enzyme
and cGMP. Nature 357:416, 1992 11. Miki N, Baraban JM, Keins JJ et al: Purification and properties of the light-activated cyclic nucleotide phosphodiesterase
of rod outer segments. J Biol Chem 260:6320, 1975 12. Palczewski K, McDowell JH, Jakes S et al: Regulation of rhodopsin dephosphorylation by arrestin. J Biol Chem 264:15770, 1989 13. Lagnado L, Baylor D: Signal flow in visual transduction. Neuron 8:995, 1992 14. Koutalos Y, Nakatani K, Yau K-W: Guanylate cyclase studied in truncated rod outer segments. Biophys J 61:428, 1992 15. Torre V, Matthews HR, Lamb TD: Role of calcium in regulating the cyclic GMP cascade of phototransduction
in retinal rods. Proc Natl Acad Sci USA 83:7109, 1986 16. Kawamura S: Rhodopsin phosphorylation as a mechanism of cyclic GMP phosphodiesterase
regulation by S-modulin. Nature 362:855, 1993 17. Yau K-W: Phototransduction mechanism in retinal rods and cones. Invest Ophthalmol Vis Sci 35:9, 1994 18. Udovichenko IP, Cunnick J, Gonzalez K, Takemoto DJ: Functional effect of phosphorylation of the photoreceptor phosphodiesterase
inhibitory subunit by protein kinase C. J Biol Chem 269:9850, 1994 19. Nathans J, Thomas D, Hogness DS: Molecular genetics of human color vision: The genes encoding blue, green, and
red pigments. Science 232:193, 1986 20. Lerea CL, Somers DE, Hurley JB et al: Isolation of specific transducin α subunits in retinal rod and cone
photoreceptors. Science 243:77, 1986 21. Lerea CL, Bunt-Milam AH, Hurley JB: α Transducin is present in blue-, green-, and red-sensitive cone photoreceptors
in the human retina. Neuron 3:367, 1989 22. Gillespie PE, Beavo JA: Characterization of a bovine cone photoreceptor phosphodiesterase purified
by cyclic GMP-sepharose chromatography. J Biol Chem 263:6122, 1988 23. Hamilton SE, Prusti RK, Bentley JK et al: Affinities of bovine photoreceptor cGMP phosphodiesterases for rod and
cone inhibitory subunits. FEBS Lett 318:157, 1993 24. Gouras P: Retinal circuitry and its relevance to diagnostic psychophysics and electrophysiology. Curr Opin Ophthalmol 3:803, 1992 25. Dacey DM: Physiology, morphology and spatial densities of identified ganglion cells
in primate retina. Ciba Found Symp 184:12, 1994 26. Kolb H: The architecture of functional neural circuits in the vertebrate retina. Invest Ophthalmol Vis Sci 35:2385, 1994 27. Bush RA, Sieving PA: A proximal retinal component in the primate photopic ERG a-wave. Invest Ophthalmol Vis Sci 35:635, 1994 28. Sieving PA, Fishman LJ, Steinberg RH: Scotopic threshold response of proximal retina in cat. J Neurophysiol 56:1049, 1986 29. Yamamoto S, Gouras P, Lopez R: The focal cone electroretinogram. Vision Res 35:1641, 1995 30. Sandberg MA, Ariel M: A hand-held two channel stimulator-ophthalmoscope. Arch Ophthalmol 101:232, 1978 31. Sutter EE, Tran D: The field topography of ERG components in man—I. Vision Res 32:443, 1992 32. Pugh ENJ, Lamb TD: Amplification and kinetics of the activation steps in phototransduction. Biochem Biophys Acta 1141:111, 1993 33. Breton ME, Schueller AW, Lamb ED, Pugh JEN: Analysis of ERG a-wave amplification and kinetics in terms of the G-protein
cascade of phototransduction. Invest Ophthalmol Vis Sci 35:295, 1994 34. Hood DC, Birch DG: A quantitative measure of the electrical activity of human rod photoreceptors
using electroretinography. Vis Neurosci 5:379, 1990 35. Thirkill CE: Cancer associated retinopathy: The CAR syndrome. Neuro Ophthalmol 13:297, 1994 36. Polans AS et al: Recoverin, a photoreceptor-specific calcium-binding protein, is expressed
by the tumor of a patient with cancer-associated retinopathy. Proc Natl Acad Sci USA 92:9176, 1995 37. Berson EL, Lessell S: Paraneoplastic night blindness with malignant melanoma. Am J Ophthalmol 106:307, 1988 38. Milan AH, Saari JC, Jacobson SG et al: Autoantibodies against retinal bipolar cells in cutaneous melanomaassociated
retinopathy. Invest Ophthalmol Vis Sci 34:91, 1933 39. Jacobson DM, Thirkill CE: Paraneoplastic cone dysfunction: An unusual visual remote effect of cancer. Arch Ophthalmol 113:1580, 1995 40. Pearson P et al: The status of Online Mendelian Inheritance in Man (OMIM) medio 1994. Nucleic Acids Res 22:3470, 1994 41. McKusick VA, Francomano CA, Antonarakis SE, Pearson PL: Mendelian Inheritance
in Man. Baltimore, The Johns Hopkins University Press, 1994 42. Heckenlively JR: Retinitis Pigmentosa. Philadelphia, JB Lippincott, 1988 43. Humphries P, Kenna P, Farrar J: On the molecular genetics of retinitis pigmentosa. Science 256:804, 1992 44. Berson EL: Retinitis pigmentosa: The Friedenwald Lecture. Invest Ophthalmol Vis Sci 34:1655, 1993 45. Rosenfeld PJ, Dryja TP: In Wiggs J (ed): Molecular Genetics of Ocular Diseases, p 99. New
York, Wiley-Liss, 1995 46. Dryja TP, Berson EL: Retinitis pigmentosa and allied diseases: Implications of genetic heterogeneity. Invest Ophthalmol Vis Sci 36:1197, 1995 47. Bunker CH, Berson EL, Bromley WC et al: Prevalence of retinitis pigmentosa in Maine. Am J Ophthalmol 97:357, 1984 48. Berson EL, Gouras P, Gunkel RD: Rod responses in retinitis pigmentosa, dominantly inherited. Arch Ophthalmol 80:355, 1968 49. Berson EL, Gouras P, Hoff M: Temporal aspects of the electroretinogram. Arch Ophthalmol 81:207, 1969 49a. Wilson EB: The sex chromosomes. Arch Mikrosk Anat 77:249, 1911 50. Haldane JBS, Sprunt AD, Haldane NM: Reduplication in mice. J Genet 5:133, 1915 51. Bird AC: Retinal photoreceptor dystrophies: The LI Edward Jackson Memorial Lecture. Am J Ophthalmol 119:543, 1995 52. Dryja TP, Li T: Molecular genetics of retinitis pigmentosa. Hum Mol Genet 4:1739, 1995 53. Nussbaum RL, Lesko JG, Lewis RA et al: Isolation of anomalous DNA sequences from within a submicroscopic X chromosomal
deletion in a patient with choroideremia, deafness, and mental
retardation. Proc Natl Acad Sci USA 84:6521, 1987 54. Cremers FPM et al: Chromosomal jumping from the DXS165 locus allows molecular characterization
of four microdeletions and a de novo chromosome X/13 translocation
with choroideremia. Proc Natl Acad Sci USA 86:7510, 1989 55. Cremers FPM, van de Pol DJR, van Kerkoff LPM et al: Cloning of a gene that is rearranged in patients with choroideraemia. Nature 347:674, 1990 56. Sankila E-M, Tolvanen R, van den Hurk JAJM et al: Aberrant splicing of the CHM gene is a significant cause of choroideremia. Nature Genet 1:109, 1992 57. Seabra MC, Brown MS, Goldstein JL: Retinal degeneration in choroideremia: Deficiency of Rab geranylgeranyltransferase. Science 259:377, 1993 58. Zhu D et al: Microdeletion in the X-chromosome and prenatal diagnosis in a family with
Norrie disease. Am J Med Genet 33:485, 1989 59. Berger W et al: Isolation of a candidate gene for Norrie disease by positional cloning. Nature Genet 1:199, 1992 60. Chen Z-Y et al: Isolation and characterization of a candidate gene for Norrie disease. Nature Genet 1:204, 1992 61. Wong F, Goldberg MF, Hao Y: Identification of a nonsense mutation at codon 128 of the Norrie's
disease gene in a male infant. Arch Ophthalmol 111:1553, 1993 62. Chen Z-Y et al: A mutation in the Norrie disease gene (NDP) associated with X-linked familial
exudative vitreoretinopathy. Nature Genet 5:180, 1993 63. Criswick VG, Schepens CL: Familial exudative vitreoretinopathy. Am J Ophthalmol 68:578, 1969 64. Stone EM et al: Genetic linkage of autosomal dominant neovascular inflammatory vitreoretinopathy
to chromosome 11q13. Hum Mol Genet 1:685, 1992 65. Kwitek-Black AE et al: Linkage of Bardet-Biedl syndrome to chromosome 16q and evidence for non-allelic
genetic heterogeneity. Nature Genet 5:392, 1993 66. Warburg M, Sjo O, Tranebjaerg L, Fledelius HC: Deletion mapping for a retinal cone-rod dystrophy: Assignment to 18q21.1. Am J Med Genet 39:288, 1991 67. Evans K et al: Genetic linkage of cone-rod retinal dystrophy to chromosome 19q and evidence
for segregation distortion. Nature Genet 6:210, 1994 68. Knowles JA et al: Identification of a locus, distinct from RDS-peripherin, for autosomal
recessive retinitis pigmentosa on chromosome 6p. Hum Mol Genet 3:14401, 1994 69. McWilliam P et al: Autosomal dominant retinitis pigmentosa (ADRP): Localization of an ADRP
gene to the long arm of chromosome 3. Genomics 5:619, 1989 70. Dryja TP et al: A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature 343:364, 1990 71. Farrar GJ et al: Autosomal dominant retinitis pigmentosa: A novel mutation in the rhodopsin
gene in the original 3q family. Hum Mol Genet 1:769, 1992 72. Farrar GJ et al: Autosomal dominant retinitis pigmentosa: Absence of the rhodopsin proline-to-histidine
substitution (codon 23) in pedigrees from Europe. Am J Hum Genet 47:941, 1990 73. Sandberg MA, Weigel-Di Franco C, Dryja TP, Berson EL: Clinical expression correlates with location of rhodopsin mutation in dominant
retinitis pigmentosa. Invest Ophthalmol Vis Sci 36:1934, 1995 74. Dryja TP et al: Mutations in the gene encoding the alpha subunit of the rod cGMP-gated
channel in autosomal recessive retinitis pigmentosa. Proc Natl Acad Sci USA 92:10177, 1995 75. Yau K-W: Cyclic nucleotide-gated channels: An expanding new family of ion channels. Proc Natl Acad Sci USA 91:3481, 1994 75a. Simell O, Takki K: Raised plasma ornithine and gyrate atrophy of the choroid and retina. Lancet 1:1031, 1973 76. Dolph PJ et al: Arrestin function in inactivation of G protein-coupled receptor rhodopsin
in vivo. Science 260:1910, 1993 77. Ramesh V et al: The ornithine aminotransferase (OAT) locus: Analysis of RFLP in gyrate
atrophy. Am J Hum Genet 42:365, 1988 78. Mitchell GA et al: Human ornithine-delta-aminotransferase: cDNA cloning and analysis of the
structural gene. J Biol Chem 263:14288, 1988 79. Inana G et al: Molecular cloning of human ornithine aminotransferase mRNA. Proc Natl Acad Sci USA 83:1203, 1986 80. Akaki Y et al: A deletion in the ornithine aminotransferase gene in gyrate atrophy. J Biol Chem 267:12950, 1992 81. Brody LC et al: Ornithine delta-aminotransferase mutations in gyrate atrophy: Allelic heterogeneity
and functional consequences. J Biol Chem 267:3302, 1992 82. Wang T et al: Mice lacking ornithine-delta-amino-transferase have paradoxical neonatal
hypoornithinaemia and retinal degeneration. Nature Genet 11:185, 1995 83. Keeler CE: The inheritance of a retinal abnormality in white mice. Proc Natl Acad Sci USA 10:329, 1924 84. Pittler SJ, Keeler CE, Sidman RL, Baehr W: PCR analysis of DNA from 70-year-old sections of rodless retina demonstrates
identity with the mouse rd defect. Proc Natl Acad Sci USA 90:9616, 1993 85. Sidman RL, Green MC: Retinal degeneration in the mouse: Location of the rd locus in linkage
group XVII. J Hered 56:23, 1965 86. Wimer RE, Wimer CC, Alameddine L, Cohen AJ: The mouse gene retinal degeneration (rd) may reduce the number of neurons
present in the adult hippocampal dentate gyrus. Brain Res 547:279, 1991 87. Lolley RN, Rong H-M, Craft CM: Linkage of photoreceptor degeneration by apoptosis with inherited defect
in phototransduction. Invest Ophthalmol Vis Sci 35:358, 1994 88. Chang C-Q, Hao Y, Wong F: Apoptosis: Final common pathway of photoreceptor death in rd, rds, and
rhodopsin mutant mice. Neuron 11:595, 1993 89. Poetera-Cailliau C, Sung CH, Nathans J, Adler R: Apoptotic photoreceptor cell death in mouse models of retinitis pigmentosa. Proc Natl Acad Sci USA 91:974, 1994 90. Schmidt SY, Lolley RN: Cyclic-nucleotide phosphodiesterase: An early defect in inherited retinal
degeneration of C3H mice. J Cell Biol 57:117, 1973 91. Farber DB, Lolley RN: Cyclic guanosine monophosphate: Elevations in degenerating photoreceptor
cells of the C3H mouse retina. Science 186:449, 1974 92. Bowes C et al: Retinal degeneration in the rd mouse is caused by a defect in the β subunit
of rod cGMP phosphodiesterase. Nature 347:677, 1990 93. Danciger M, Bowes C, Kozak CA et al: Fine mapping of a putative rd cDNA and its cosegregation with rd expression. Invest Ophthalmol Vis Sci 31:1427, 1990 94. Pittler SJ, Baehr W: Identification of a nonsense mutation in the rod photoreceptor cGMP phosphodiesterase
beta-subunit gene of the rd mouse. Proc Natl Acad Sci USA 88:8322, 1991 95. Farber DB, Danciger J, Aguirre G: The beta subunit of cyclic GMP phosphodiesterase mRNA is deficient in canine
rod-cone dysplasia. Neuron 9:349, 1992 96. Suber ML et al: Irish setter dogs affected with rod/cone dysplasia contain a nonsense mutation
in the rod cGMP phosphodiesterase beta-subunit gene. Proc Natl Acad Sci USA 90:3968, 1993 97. Ray K, Baldwin VJ, Acland GM et al: Cosegregation of codon 807 mutation of the canine rod cGMP phosphodiesterase
beta gene and rcd1. Invest Ophthalmol Vis Sci 35:4291, 1994 98. Riess O, Noerremoelle A, Weber B et al: The search for mutations in the gene for the beta subunit of the cGMP phosphodiesterase (PDEB) in
patients with autosomal recessive retinitis
pigmentosa. Am J Hum Genet 51:755, 1992 99. McLaughlin ME, Sandberg MA, Berson EL, Dryja TP: Recessive mutations in the gene encoding the beta-subunit of rod phosphodiesterase
in patients with retinitis pigmentosa. Nature Genet 4:130, 1993 100. McLaughlin ME, Ehrhart TL, Berson EL, Dryja TP: Mutation spectrum of the gene encoding the beta subunit of rod phosphodiesterase
among patients with autosomal recessive retinitis pigmentosa. Proc Natl Acad Sci USA 92:3249, 1995 101. Bayes M et al: Homozygous tandem duplication within the gene encoding the beta-subunit
of rod phosphodiesterase as a cause for autosomal recessive retinitis
pigmentosa. Hum Mutat 5:228, 1995 102. Gal A, Orth U, Baehr W et al: Heterozygous missense mutation in the rod cGMP phosphodiesterase beta-subunit
in autosomal dominant stationary night blindness. Nature Genet 7:64, 1994 103. Huang SH et al: A mutation in the gene encoding the alpha-subunit of rod cGMP phosphodiesterase (PDEA) in
retinitis pigmentosa. Invest Ophthalmol Vis Sci 36(suppl):S825, 1995 104. Gibson F et al: A type VII myosin encoded by the mouse deafness gene shaker-1. Nature 374:62, 1995 105. Hasson T, Heintzelman MB, Santos-Sacchi J et al: Expression in cochlea and retina of myosin VIIa, the gene product defective
in Usher syndrome type 1B. Proc Natl Acad Sci USA 92:9815, 1995 106. Weil D et al: Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature 374:60, 1995 107. Gardner JM et al: The mouse pink-eye dilution gene: Association with human Prader-Willi and
Angelman syndromes. Science 257:1121, 1992 108. Rinchik EM et al: A gene for the mouse pink-eye dilution locus and for human type II oculocutaneous
albinism. Nature 361:72, 1993 109. Lee S-T et al: Mutations of the P gene in oculocutaneous albinism, ocular albinism and
Prader-Willi syndrome plus albinism. N Engl J Med 330:529, 1994 110. Travis GH, Brennan MB, Danielson PE et al: Identification of a photoreceptor-specific mRNA encoded by the gene responsible
for retinal degeneration slow (rds). Nature 338:70, 1989 111. Ma J et al: Retinal degeneration slow (rds) in mouse results from simple insertion
of a t haplotype-specific element into protein-coding exon II. Genomics 28:212, 1995 112. Travis GH, Sutcliffe JG, Bok D: The retinal degeneration slow (rds) gene product is a photoreceptor disc
membrane-associated glycoprotein. Neuron 6:61, 1991 113. Travis GH et al: The human retinal degeneration slow (RDS) gene: Chromosomal assignment
and structure of the mRNA. Genomics 10:733, 1991 114. Farrar GJ et al: A three-base-pair deletion in the peripherin-RDS gene in one form of retinitis
pigmentosa. Nature 354:478, 1991 115. Kajiwara K et al: Mutations in the human retinal degeneration slow gene in autosomal retinitis
pigmentosa. Nature 354:480, 1991 116. Bascom RA et al: Localization of the photoreceptor gene ROM1 to human chromosome 11 and
mouse chromosome 19: Sublocalization of human 11q13 between PGA and PYGM. Am J Hum Genet 51:1028, 1992 117. Kajiwara K, Berson EL, Dryja TP: Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS
and ROM1 loci. Science 264:1604, 1994 118. Hill RE et al: Mouse small eye results from mutations in a paired-liked homeobox-containing
gene. Nature 354:522, 1991 119. Glaser T et al: PAX gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia, and
central nervous system defects. Nature Genet 7:463, 1994 120. Ton CCT et al: Positional cloning and characterization of a paired box- and homeobox-containing
gene from the aniridia region. Cell 67:1059, 1991 121. Hanson IM et al: Mutations at the PAX6 locus are found in heterogeneous anterior segment
malformations including Peters' anomaly. Nature Genet 6:168, 1994 122. Mirzayans F, Pearce WG, MacDonald IM, Walter MA: Mutation of the PAX6 gene in patients with autosomal dominant keratitis. Am J Hum Genet 57:539, 1995 123. Bergen AAB, ten Brink JB, Riemslang F et al: Localization of a novel X-linked congenital stationary night blindness
locus: Close linkage to the RP3 type retinitis pigmentosa gene region. Hum Mol Genet 4:931, 1995 124. Dryja TP, Berson EL, Rao VR, Oprian DD: Heterozygous missense mutation in the rhodopsin gene as a cause of congenital
stationary night blindness. Nature Genet 4:280, 1993 125. Sieving PA et al: Dark-light: Model for nightblindness from the human rhodopsin gly90-to-asp
mutation. Proc Natl Acad Sci USA 92:880, 1995 126. Rao V, Cohen GB, Oprian DB: Rhodopsin mutation G90D and a molecular mechanism for congenital night
blindness. Nature 367:639, 1994 127. Fuchs S et al: A homozygous 1-base pair deletion in the arrestin gene is a frequent cause
of Oguchi disease in Japanese. Nature Genet 10:360, 1995 128. Small KW: In Wiggs J (ed): Molecular Genetics of Ocular Diseases, p 127. New
York, Wiley-Liss, 1995 129. Small KW et al: North Carolina macular dystrophy is assigned to chromosome 6. Genomics 13:681, 1992 130. Nichols BE et al: Refining the locus for Best vitelliform macular dystrophy and mutation
analysis of the candidate gene ROM1. Am J Hum Genet 54:95, 1994 131. Deutman AF, Pickers AJL, Aan de Kerk AL: Dominantly inherited cystoid macular edema. Am J Ophthalmol 82:540, 1976 132. Fishman GA, Goldberg MF, Trautmann JC: Dominantly inherited cystoid macular edema. Ann Ophthalmol 11:21, 1979 133. Kremer H et al: Localization of the gene for dominant cystoid macular dystrophy on chromosome 7p. Hum Mol Genet 3:299, 1994 134. Weber BHF, Vogt G, Wolz W et al: Sorsby's fundus dystrophy is genetically linked to chromosome 22q13-ter. Nature Genet 7:158, 1994 135. Weber BHF, Vogt G, Pruett RC et al: Mutations in the tissue inhibitor metalloproteinases-3 (TIMP3) in patients
with Sorsby's fundus dystrophy. Nature Genet 8:352, 1994 136. Jacobson SG et al: Night blindness in Sorsby's fundus dystrophy reversed by vitamin A. Nature Genet 11:27, 1995 137. Wells J et al: Mutations in the human retinal degeneration slow (RDS) gene can cause either
retinitis pigmentosa or macular dystrophy. Nature Genet 3:213, 1993 138. Nichols BE et al: Butterfly-shaped pigment dystrophy of the fovea caused by a point mutation
in codon 167 of the RDS gene. Nature Genet 3:202, 1993 139. Nichols BE et al: A 2 base pair deletion in the RDS gene associated with butterfly-shaped
pigment dystrophy of the fovea. Hum Mol Genet 2:601, 1993 140. Feist RM, White MF Jr, Skalka H, Stone EM: Choroidal neovascularization in a patient with adult foveomacular dystrophy
and a mutation in the retinal degeneration slow gene (pro210-to-arg). Am J Ophthalmol 118:259, 1994 141. Kim RY et al: Autosomal dominant pattern dystrophy of the retina associated with a 4-base
pair insertion at codon 140 in the peripherin/RDS gene. Arch Ophthalmol 113:451, 1995 142. Kikawa E, Nakazawa M, Chida Y et al: A novel mutation (asn244-to-lys) in peripherin/RDS gene causing autosomal
dominant retinitis pigmentosa associated with bull's-eye maculopathy
detected by nonradioisotopic SSCP. Genomics 20:137, 1994 143. Kajiwara K, Sandberg MA, Berson EL, Dryja TP: A mutation in the human peripherin/RDS gene in a family with autosomal
dominant retinitis punctata albescens. Nature Genet 3:208, 1993 144. Olschwang S et al: Restriction of ocular fundus lesions to a specific subgroup of APC mutations
in adenomatous polyposis coli patients. Cell 75:959, 1993 145. Moraes CT et al: Mitochondrial DNA deletions in progressive external ophthalmoplegia and
Kearns-Sayer syndrome. N Engl J Med 320:1293, 1989 146. Holt IJ, Harding AE, Petty RKH, Morgan-Hughes JA: A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am J Hum Genet 46:428, 1990 147. Wallace DC et al: Mitochondrial DNA mutation associated with Leber hereditary optic neuropathy. Science 242:1427, 1988 148. Stone EM et al: Visual recovery in patients with Lever's hereditary optic neuropathy
and the 11778 mutation. J Clin Neuro Ophthalmol 12:10, 1992 149. Min KC, Zvyaga TA, Cypess AM, Sakmar TP: Characterization of mutant rhodopsins responsible for autosomal dominant
retinitis pigmentosa: Mutations on the cytoplasmic surface affect transducin
activation. J Biol Chem 1993:9400, 1993 150. Noell WK: Possible mechanisms of photoreceptor damage by light in mammalian eyes. Vision Res 20:1163, 1980 151. Fain GL, Lisman JE: Photoreceptor degeneration in vitamin A deprivation and retinitis pigmentosa: The
equivalent light hypothesis. Exp Eye Res 57:335, 1993 152. Li T, Franson WK, Gordon JW et al: Constitutive activation of phototransduction by K296E opsin is not a cause
of photoreceptor degeneration. Proc Natl Acad Sci USA 92:3551, 1995 153. Robinson PR, Cohen GB, Zhukovsky EA, Oprian DD: Constitutively active mutants of rhodopsin. Neuron 9:719, 1992 154. Robinson PR, Buczylko J, Ohguro H, Palczewski K: Opsins with mutations at the site of chromophore attachment constitutively
activate transducin but are not phosphorylated by rhodopsin kinase. Proc Natl Acad Sci USA 91:5411, 1994 155. Raport CJ et al: Downregulation of cGMP phosphodiesterase induced by expression of GTPase-deficient
cone transducin in mouse rod photoreceptors. Invest Ophthalmol Vis Sci 35:2932, 1994 156. Chen J, Makino CL, Peachey NS et al: Mechanisms of rhodopsin inactivation in vivo as revealed by a COOH-terminal
truncation mutant. Science 267:374, 1995 157. Naash MI, Hollyfield JG, al-Ubaidi MR, Baehr W: Simulation of human autosomal dominant retinitis pigmentosa in transgenic
mice expressing a mutated murine opsin gene. Proc Natl Acad Sci USA 90:5499, 1993 158. Olson JE et al: Transgenic mice with a rhodopsin mutation (Pro23His): A mouse model of
autosomal dominant retinitis pigmentosa. Neuron 9:815, 1992 159. Roof DJ, Adamian M, Hayes A: Rhodopsin accumulation at abnormal sites in retinas of mice with a human
P23H rhodopsin transgene. Invest Ophthalmol Vis Sci 35:4049, 1994 160. Sung CH, Makino C, Baylor D, Nathans J: A rhodopsin gene mutation responsible for autosomal dominant retinitis
pigmentosa results in a protein that is defective in localization to the
photoreceptor outer segment. J Neurosci 14:5818, 1994 161. Capecchi M: The new mouse genetics: Altering the genome by gene targeting. Trends Genet 5:70, 1989 162. Hogan B, Beddington R, Costantini F, Lacy E: Manipulating the Mouse Embryo: A
Laboratory Manual. Cold Spring Harbor, NY, Cold Spring Harbor Laboratory
Press, 1994 163. Flannery JG, Chen J, Xu J, Simon MI: Overexpression of Bcl-2 can interfere with apoptosis in retinal degeneration. Invest Ophthalmol Vis Sci 36(suppl 4):S615, 1995 |