MICROPHTHALMOS AND ANOPHTHALMOS
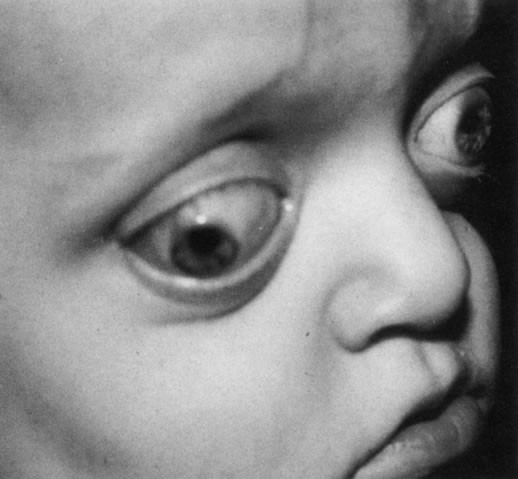
True anophthalmos is a rare condition that results from failure of development or complete regression of the optic vesicle. This condition may be clinically indistinguishable from severe microphthalmos, which results from incomplete invagination of the optic vesicle or closure of the embryonic fissure. The term clinical anophthalmos has been used to describe patients who have no clinical or radiographic evidence of any ocular remnant, although true anophthalmos can only be verified after careful histologic sectioning of the orbital tissues.
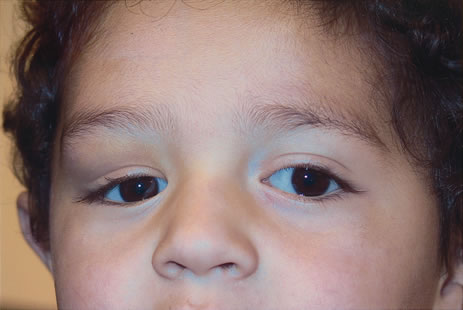
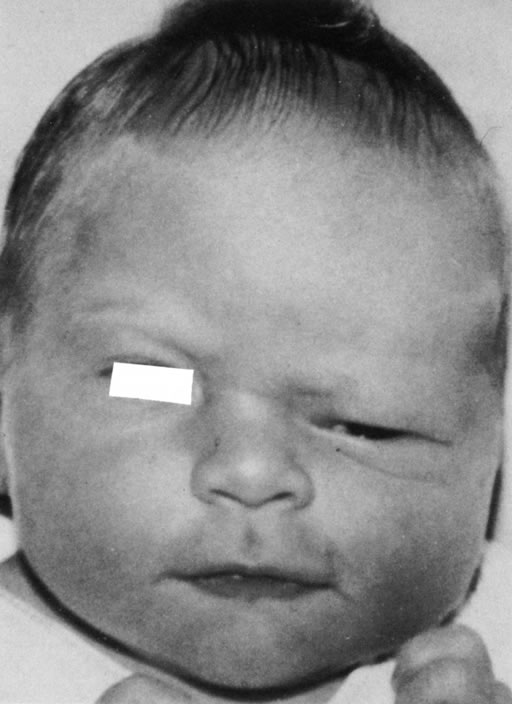
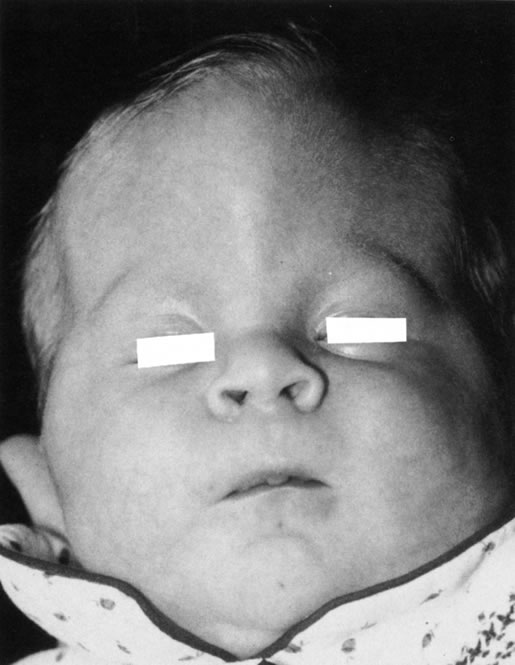
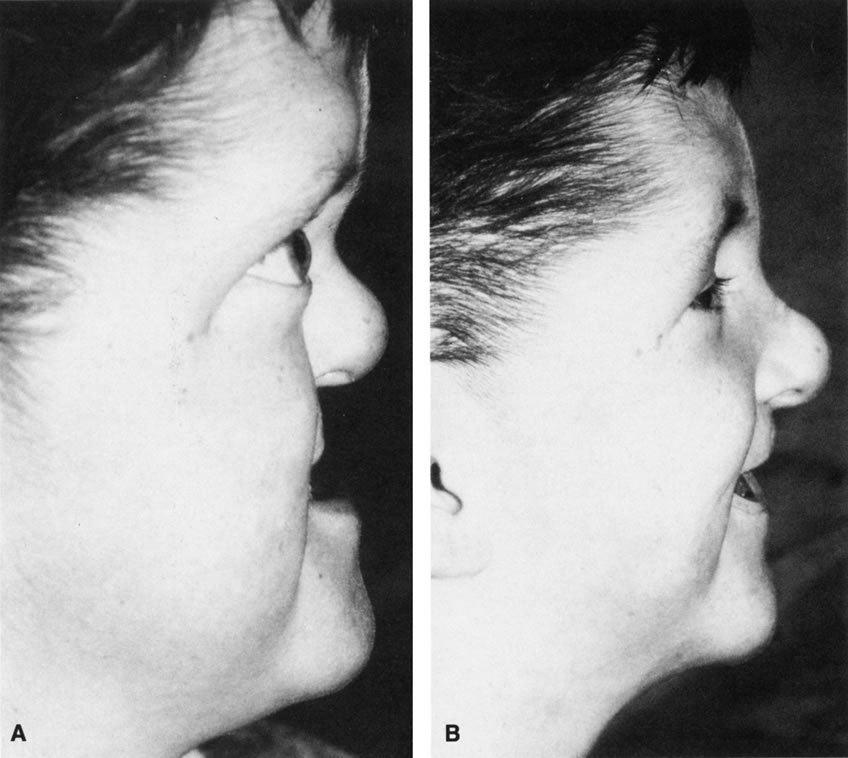
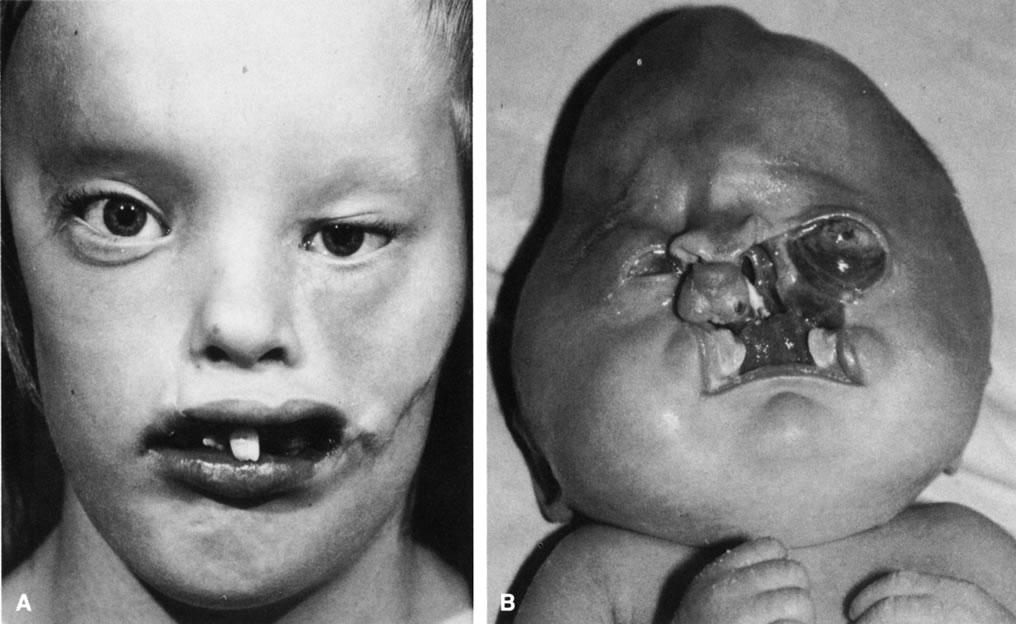
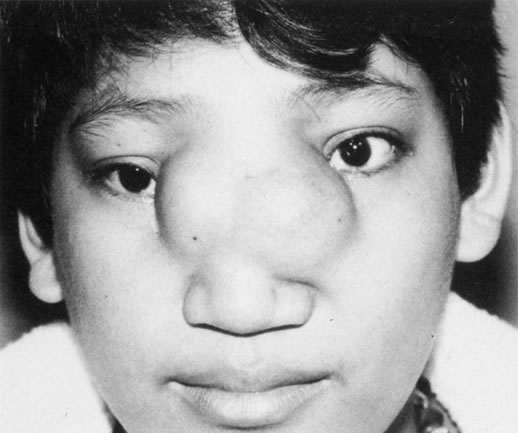
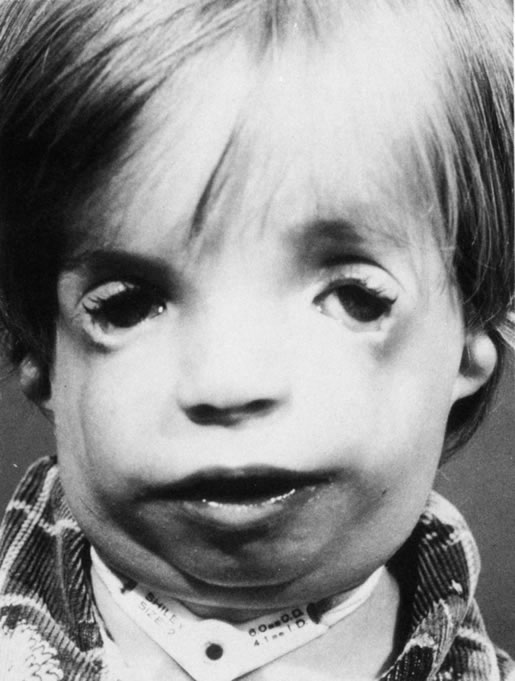
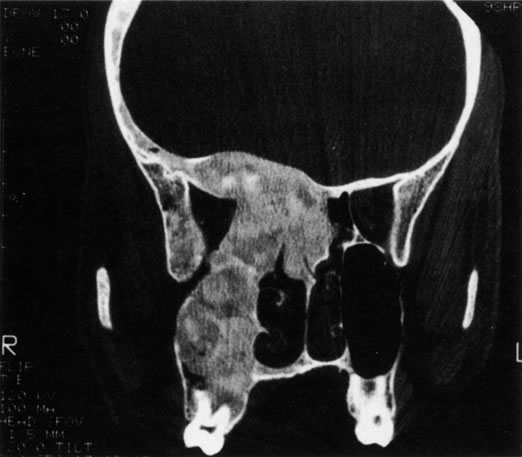
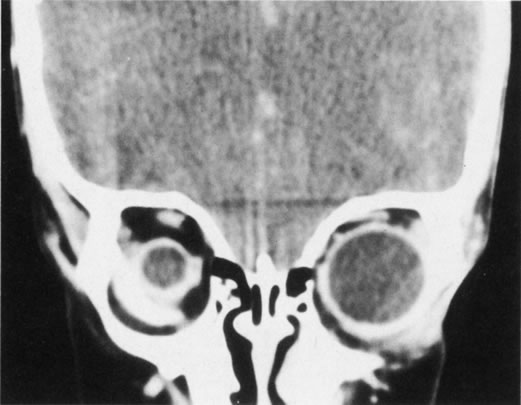
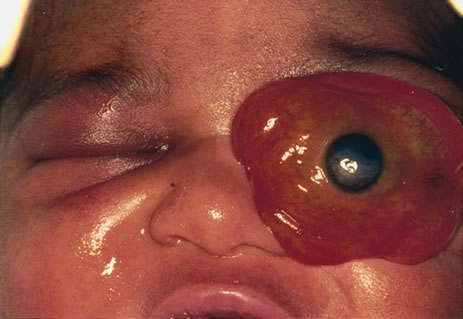
Anophthalmos and microphthalmos are usually unilateral and may be associated with a variety of craniofacial and systemic anomalies, including orbital hypoplasia, facial clefts, basal encephalocele, hemifacial microsomia, mandibulofacial dysostosis, cardiac anomalies, polydactyly, and mental retardation. When they occur unilaterally, they also can be associated with anomalies of the contralateral “normal” eye, including cataract, cornea1 opacities, microphthalmos, coloboma, epibulbar dermoids, and nystagmus. Anophthalmos and severe microphthalmos frequently are associated with contracted conjunctival fornices, phimotic eyelids, and generalized hypoplasia of the periocular soft tissues (Fig. 1). When soft tissue contractures occur, the early use of conformers is essential to expand these tissues.2 This treatment should be instituted in the first month of life, with progressive enlargement of the conformer over time to achieve maximum expansion of the conjunctival fornix. Unfortunately, this treatment usually does not stimulate adequate orbital bone growth, and unilateral microphthalmos and anophthalmos may be associated with secondary orbital hypoplasia (Fig. 2). Serial implantation of progressively larger orbital implants or placement of expansile orbital implants has been advocated to stimulate bony orbital development.3,4
|
|
CYSTIC ANOMALIES OF THE ORBIT
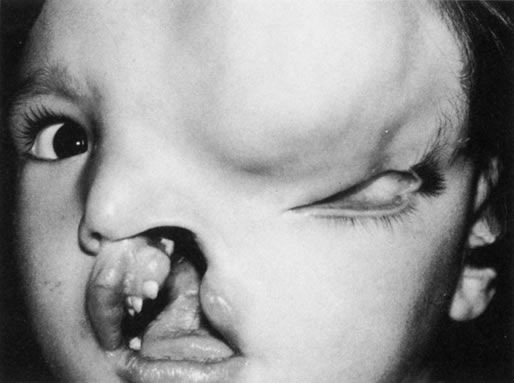
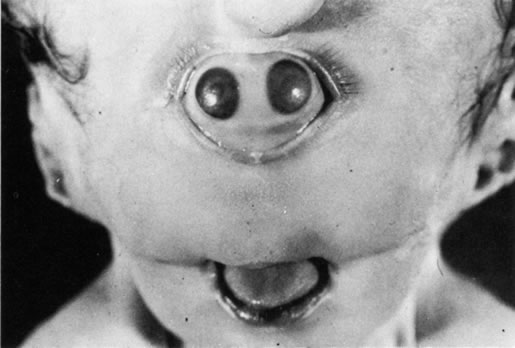
Many congenital cystic structures may arise from or involve the orbit. Some cystic structures, such as meningoencephaloceles or mucoceles, result from defects in the bony sutures of the cranial skeleton, allowing herniation of adjacent structures into the orbit. Other cystic structures, such as dermoid cysts, teratomas, and epithelial cysts, result from developmental anomalies of the orbital soft tissues. Most isolated orbital cysts have a subtle clinical presentation at birth, although some may present with extreme proptosis (Fig. 3). Ultrasonography can aid in the prenatal detection and monitoring of large orbital cysts.5
|
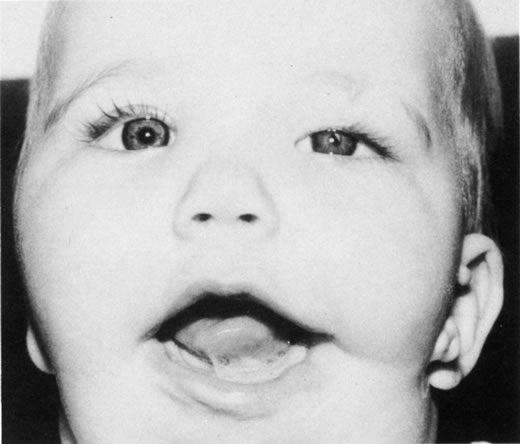
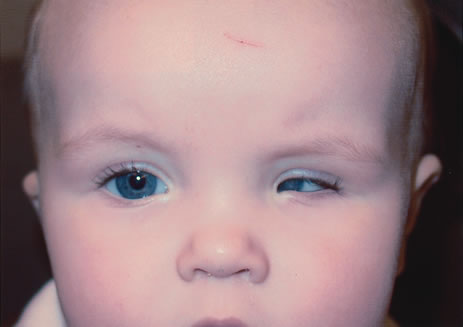
Dermoid cysts are the most common congenital orbital anomalies and represent developmental choristomas that are believed to arise from ectodermal nests pinched off by the fusion of bony sutures around the orbit. These cysts often originate from the frontozygomatic suture temporally but can also be seen nasally, arising from the frontonasal and frontolacrimal sutures; they rarely occur deep in the orbit.6 They commonly present during the first decade of life as a well-circumscribed, firm, rubbery subcutaneous mass just below the temporal eyebrow. Deeper dermoids can remain asymptomatic for many years, often presenting later in life as a slowly expanding orbital mass. Complete excision of these encapsulated lesions is the preferred treatment. Rupture of the cyst from trauma or during surgery may result in severe orbital inflammation (Fig. 4).
|