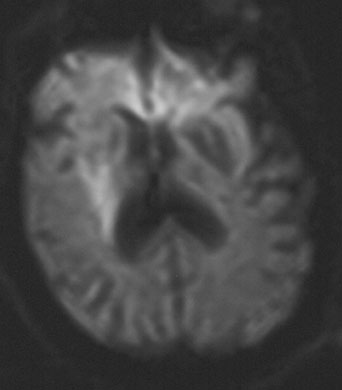
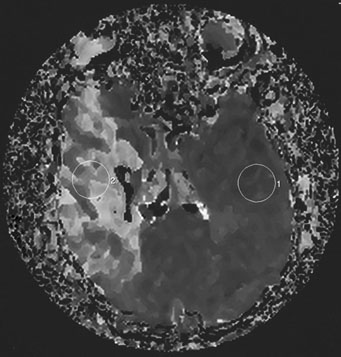
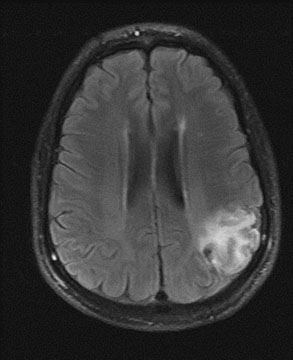
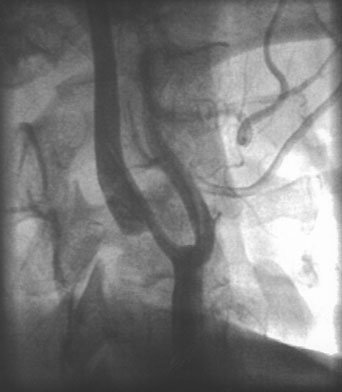
1. American Heart Association: 2004 Heart and Stroke Statistics—2004 Update. Dallas, TX (http://www.americanheart.org) 2. Taylor TN, Davis PH, Torner JC, et al: Lifetime cost of stroke in the united States. Stroke 27:1459, 1996 3. Barba R, Martínez-Espinosas S, Rodriguez-García E, et al: Post-stroke dementia: clinical features and risk factors. Stroke 31:1494, 2000 4. Prencipe M, Ferretti C, Casini AR, et al: Stroke, disability, and dementia: results of a population survey. Stroke 28:531, 1997 5. Astrom M, Adolfsson R, Asplund K. Major depression in stroke patients: a 3 year longitudinal study. Stroke 24:976, 1993 6. Wilkinson PR, Wolfe CDA, Warburton FG, et al: A long-term follow-up of stroke patients. Stroke 28:507, 1997 7. US Bureau of the Census. Statistical abstract for the United States: 1995. 115th ed. Washington, DC: US Bureau of the Census, 1995 8. Bonita R. Epidemiology of stroke. Lancet 339:342, 1992 9. Wray SH. Visual symptoms (eye). In Bogousslavsky J, Caplan LR (eds): Stroke Syndromes. New York: Cambridge University Press, 1995 10. Furlan AJ, Whisnant JP, Kearns TP. Unilateral visual loss in bright light: an unusual symptom of carotid artery
occlusive disease. Arch Neurol 36:675, 1979 11. Saver JL, Biller J. Superficial middle cerebral artery. In Bogousslavsky J, Caplan LR (eds): Stroke Syndromes. New York: Cambridge University Press, 1995 12. Bogousslavsky J, Regli F. Anterior cerebral artery territory infarction in the Lausanne Stroke Registry: clinical
and etiological patterns. Arch Neurol 47:144, 1990 13. Helgason C, Caplan LR, Goodwin J, et al: Anterior choroidal artery territory infarction. Arch Neurol 43:681, 1986 14. Caplan LR, Amarenco P, Rosengart A, et al: Embolism from vertebral artery origin disease. Neurology 45:1505, 1992 15. Caplan LR. Top of the basilar syndrome. Neurology 30:72, 1980 16. Bogousslavsky J, Moulin T. Border-zone infarcts. In Bogousslavsky J, Caplan LR (eds): Stroke Syndromes. New York: Cambridge University Press, 995:358–365 17. Sage JI. “Man-in-the-barrel syndrome” after cerebral hypoperfusion: clinical
description, incidence and prognosis. Ann Neurol 14:131, 1983 18. Graeber MC, Jordan JE, Mishra SK, et al: Watershed infarction on computed tomographic scan: an unreliable sign of
hemodynamic stroke. Arch Neurol 49:311, 1992 19. Mitsias P. Head pain and stroke. In Welch KMA, Caplan LR, Reis DJ, Siesjo BK, Weir B (eds): Primer on Cerebrovascular Diseases. New York: Academic Press, 1997 20. Fisher CM. Lacunar strokes and infarcts. A review. Neurology 32:871, 1982 21. Fisher CM. Binswanger's encephalopathy: a review. J Neurol 236:65, 1989 22. Tatemichi TK. Dementia. In Bogousslavsky J, Caplan LR (eds): Stroke Syndromes. New York: Cambridge University Press, 1995:161–181 23. Ameri A, Bousser MG. Cerebral venous thrombosis. Neurol Clin 10:87, 1992 24. Easton JD, Albers GW, Caplan LR, et al: TIA Working Group. Reconsideration of TIA terminology and definitions. Neurology 62(8 Suppl 6):S29, 2004 25. Dyken ML, Conneally M, Haerer AF, et al: Cooperative study of hospital frequency and character of transient ischemic
attacks. I. Background, organization, and clinical survey. JAMA 237: 882, 1977 26. Levy DE. How transient are transient ischemic attacks? Neurology 38:674, 1988 27. Ay H, Oliveira-Filho J, Buonanno FS, et al: ‘Footprints’ of transient ischemic attacks: a diffusion-weighted
MRI study. Cerebrovasc Dis 14:177, 2002 28. Jones T, Morawetz R, Crowell RM, et al: Threshold of focal cerebral ischemia in awake monkeys. J Neurosurg 54:773, 1981 29. Babikian VL, Wijman C, Hyde C, et al: Cerebral microembolism and early recurrent cerebral or retinal ischemic
events. Stroke 28:1314, 1997 30. Siebler M, Kleinschmidt A, Sitzer M, et al: Cerebral microembolism in symptomatic and asymptomatic high grade internal
carotid artery stenosis. Neurology 44:615, 1994 31. Russel D. The detection of cerebral emboli using transcranial Doppler. Theoretical, experimental
and clinical aspects. In Newell DW, Aaslid R (eds): Transcranial Doppler. New York: Raven Press, Ltd., 1992:207–214. 32. Fisher CM. Concerning recurrent transient cerebral ischemic attacks. Can Med Assoc J 86:1091, 1962 33. Johnston SC, Gress DR, Browner WS, et al: Short-term prognosis after emergency department diagnosis of TIA. JAMA 284: 290, 2000 34. Rothman SM, Olney JW. Glutamate and the pathophysiology of hypoxic-ischemic brain damage. Ann Neurol 19:105, 1986 35. Castillo J, Dávalos A, Noya M. Progression of ischemic stroke and excitotoxic amino acids. Lancet 349:79, 1997 36. Adams HP, Brott TG, Furlan AJ, et al: Guidelines for thrombolytic therapy for acute stroke: a supplement to the
guidelines for the management of patients with acute ischemic stroke. Circulation 94:1167, 1996 37. The National Institute of Neurological Disorders and Stroke rt-Pa Stroke
Study Group. Tissue plasminogen activator for acute ischemic stroke. N Engl J Med 333:1581, 1995 38. van Everdingen KJ, van der Grond J, Kappelle LJ, et al: Diffusion-weighted magnetic resonance imaging in acute stroke. Stroke 29:1783, 1998 39. Lansberg MG, Albers GW, Beaulieu C, et al: Comparison of diffusion-weighted MRI and CT in acute stroke. Neurology 54:1557, 2000 40. Warach S, Chien D, Li W, et al: Fast magnetic resonance diffusion-weighted imaging of acute human stroke. Neurology 42:1717, 1992 41. Brott T, Adams HP, Olinger CP, et al: Measurements of acute cerebral infarction: a clinical examination scale. Stroke 20:864, 1989 42. Furlan A, Higashida R, Wechsler L, et al: Intra-arterial prourokinase for acute ischemic stroke. The PROACT II study: a
randomized controlled trial. Prolyse in Acute Cerebral Thromboembolism. JAMA 282:2003, 1999 43. Brandt T, von Kummer R, Muller-Kuppers M, et al: Thrombolytic therapy of acute basilar artery occlusion: variables affecting
recanalization and outcome. Stroke 27:875, 1996 44. Australian Urokinase Stroke Trial (AUST). Stroke 29:550, 1998 45. Warach S. Desmoteplase in Acute Ischemic Stroke. 29th International Stroke Conference, 2004 46. Adams HP, Adams RJ, Brott T, et al: Guidelines for the early management of patients with ischemic stroke. Stroke 34:1056, 2003 47. Martinez-Vila E, Sieira PI. Current status and perspectives of neuroprotection in ischemic stroke treatment. Cerebrovasc Dis 11:60, 2001 48. Cardiogenic Brain Embolism: The Second Report of the Cerebral Embolism
Task Force. Arch Neurol 46:727, 1989 49. The Publications Committee for the Trial of Org 10172 in acute stroke treatment (TOAST) Investigators. Low molecular weight heparinoid ORG 10172 (danaparoid), and outcome
after acute ischemic stroke: a randomized controlled trial. JAMA 279:1265, 1998 50. International Stroke Trial Collaborative Group. The International Stroke Trial (IST): a randomised trial of aspirin, subcutaneous
heparin, both, or neither among 19435 patients with
ischaemic stroke. Lancet 349:1569, 1997 51. Berge E, Abdelnoor M, Nakstad PH, et al: Low molecular weight heparin versus aspirin in patients with acute ischemic
stroke and atrial fibrillation: a double-blind randomized study. Lancet 355:1205, 2000 52. Saxena R, Lewis S, Berge E, et al: Risk of early death and recurrent stroke and effect of hepain in 3169 with
acute ischemic stroke and atrial fibrillation in the International
Stroke Trial. Stroke 32:2333, 2001 53. CAST (Chinese Acute Stroke Trial) Collaborative Group. CAST: a randomised placebo-controlled trial of early aspirin use in 20,000 patients
with acute ischaemic stroke. Lancet 349:1641, 1997 54. Bock RW, Lusby RJ. Lesions, dynamics, and pathogenetic mechanisms responsible for ischemic
events in the brain. In Moore WS (ed): Surgery for Cerebrovascular Disease. Philadelphia: WB Saunders, 1996:48–71 55. Sacco RL, Foulkes MA, Mohr JP, et al: Determinants of early recurrence of cerebral infarction: Stroke Data Bank. Stroke 20:983, 1989 56. Wolf PA, D'Agostino RB, O'Neal A, et al: Secular trends in stroke incidence and mortality: The Framingham Study. Stroke 23:1551, 1992 57. Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation: a major contributor to stroke in the elderly. The
Framingham Study. Arch Intern Med 147:1561, 1987 58. Wolf PA, Dawber TR, Thomas HE, et al: Epidemiologic assessment of chronic atrial fibrillation and risk of stroke: The
Framingham Study. Neurology 28:973, 1978 59. The Stroke Prevention in Atrial Fibrillation investigators. Predictors of thromboembolism in atrial fibrillation. I. Clinical features
of patients at risk. Ann Intern Med 116:1, 1992 60. The Stroke Prevention in Atrial Fibrillation Investigators. Predictors of thromboembolism in atrial fibrillation. II. Echocardiographic
features of patients at risk. Ann Intern Med 116:6, 1992 61. Benjamin EJ, Levy D, Vaziri SM, et al: Independent risk factors for atrial fibrillation in a population-based
cohort: The Framingham Heart Study. JAMA 271:840, 1994 62. Connolly SJ, Paupacis A, Gent M, et al: Canadian Atrial Fibrillation Anticoagulation (CAFA) Study. J Am Coll Cardiol 18:349, 1991 63. Lechat P, Mas JL, Lascault G, et al: Prevalence of patent foramen ovale in patients with stroke. N Engl J Med 318:1148, 1988 64. Mas JL, Zuber M, for the French Study Group on Patent Foramen Ovale and Atrial Septal
Aneurysm. Recurrent cerebrovascular events in patients with patent foramen ovale, atrial
septal aneursym, or both and cryptogenic stroke or transient ischemic
attack. Am Heart J 130:1083, 1995 65. Forteza AM, Koch S, Romano JG, et al: Transcranial Doppler detection of fat emboli. Stroke 30:2687, 1999 66. Knauth M, Ries S, Pohimann S, et al: Cohort study of multiple brain lesions in sport divers: role of a patent
foramen ovale. BMJ 314:701, 1997 67. DeRock FA, Comess KA, Albers G, et al: Transesophageal echocardiography in the evaluation of stroke. Ann Intern Med 117:992, 1992 68. The Atrial Fibrillation Investigators. Risk factors for stroke and efficacy of antithrombotic therapy in atrial
fibrillation: analysis of pooled data from five randomized controlled
trials. Arch Intern Med 154:1449, 1994 69. Albers GW, Easton JD, Sacco RL, et al: Antithrombotic and thrombolytic therapy for ischemic stroke. Chest 114:683s, 1998 70. Homma S, Sacco RL, Di Tullio MR, et al: PFO in Cryptogenic Stroke Study (PICSS) Investigators. Effect
of medical treatment in stroke patients with patent foramen ovale: patent
foramen ovale in Cryptogenic Stroke Study. Circulation 105:2625, 2002 71. O'Leary DH, Polak JF, Kronmal RA, et al: Carotid artery intima and media thickness as a risk factor for myocardial
infarction and stroke in older adults. N Engl J Med 340:14, 1999 72. Stary HC, Chandler AB, Dinsmore RE, et al: A definition of advanced types of atherosclerotic lesions and a histological
classification of atherosclerosis. A report from the Committee on
Vascular Lesions of the Council on Arteriosclerosis, American Heart
Association. Circulation 92:1355, 1995 73. The North American Symptomatic Carotid Endarterectomy Trial Collaborators. Beneficial effect of carotid endarterectomy in symptomatic patients with
high-grade stenosis. N Engl J Med 325:445, 1991 74. The North American Symptomatic Carotid Endarterectomy Trial Collaborators. Beneficial effect of carotid endarterectomy in patients with symptomatic
moderate or severe stenosis. N Engl J Med 339:1415, 1998 75. Executive Committee for the Asymptomatic Carotid Atherosclerosis Study. Endarterectomy for asymptomatic carotid artery stenosis. JAMA 273:1421, 1995 76. Ringelstein EB, Zeumer H, Angelou D. The pathogenesis of strokes from internal carotid artery occlusion: diagnostic
and therapeutical implications. Stroke 14:867, 1983 77. Klijn CJ, Kappelle LJ, van Huffelen AC, et al: Recurrent ischemia in symptomatic carotid occlusion: Prognostic value
of hemodynamic factors. Neurology 55:1806, 2000 78. Bisschops RHC, Klijn CJM, Kappelle LJ, et al: Collateral flow and ischemic brain lesions in patients with unilateral
carotid artery occlusion. Neurology 60:1435, 2003 79. Gomez CR. Carotid plaque morphology and risk for stroke. Stroke 21:148, 1990 80. Polak JF, Shemanski L, O'Leary D, et al: Hypoechoic plaque at US of the carotid artery: an independent risk factor
for incident stroke in adults aged 65 or older. Radiology 208:649, 1998 81. Blakeley DD, Oddone EZ, Hasselblad V, et al: Non-invasive carotid artery testing: a meta-analytic review. Ann Intern Med 122:360, 1995 82. Heiserman JE, Zabramski JM, Drayer BP, et al: Clinical significance of the flow gap in carotid magnetic resonance angiography. J Neurosurg 85:384, 1996 83. Forteza AM, Rabinstein A. Angioplastía y Stenting de la estenosis carotídea. Alternativa
terapéutica o posibilidad técnica? Rev Neurol 32:270, 2001 84. CAVATAS Investigators. Endovascular versus surgical treatment in patients with carotid stenosis
in the Carotid and Vertebral Artery Transluminal Angioplasty Study (CAVATAS): a
randomised trial. Lancet 357:1729, 2001 85. Howard VJ, Sheffet AJ, Brott TG, et al: Crest. Progress Report of the Carotid Revascularization Endarterectomy
vs. Stenting Trial (CREST). 29th International Stroke Conference, 2004 86. SAPPHIRE Investigators. Stenting and Angioplasty with Protection in Patients and High Risk for
Endarterectomy. 28th International Stroke Conference, 2003 87. Chimowitz MI, Kokkinos J, Strong J, et al: for the Warfarin-Aspirin Symptomatic Intracranial Disease Study Group. The
Warfarin-Aspirin Symptomatic Intracranial Disease Study. Neurology 45:1488, 1995 88. Adams RJ, McKie VC, Carl EM, et al: Long-term stroke risk in children with sickle cell disease screened with
transcranial Doppler. Ann Neurol 42:699, 1997 89. Adams RJ, McKie VC, Hsu L, et al: Prevention of a first stroke by transfusions in children with sickle cell
anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med 339:5, 1998 90. Goldstein LB, Adams R, Becker K, et al: Primary prevention of ischemic stroke: a statement for healthcare professionals
from the Stroke Council of the American Heart Association. Stroke 32:280, 2001 91. Amarenco P, Cohen A, Tzourio G, et al: Atherosclerotic disease of the aortic arch and the risk factor of ischemic
stroke. N Engl J Med 331:1474, 1994 92. Schievink WI. Spontaneous dissections of the carotid and vertebral arteries. N Engl J Med 344:898, 2001 93. Hicks PA, Leavitt JA, Mokri B. Ophthalmic manifestations of vertebral artery dissection: patients seen
at the Mayo Clinic from 1976 to 1992. Ophthalmology 101:1786, 1994 94. Mokri B, Schievink WI, Olsen KD, et al: Spontaneous dissection of the cervical internal carotid artery: presentation
with lower cranial nerve palsies. Arch Otolaryngol Head Neck Surg 118:431, 1992 95. Scievink WI. The treatment of spontaneous carotid and vertebral artery dissections. Curr Opin Cardiol 15:316, 2000 96. Fisher CM. Lacunes: small deep cerebral infarcts. Neurology 15:774, 1965 97. Fisher CM. The arterial lesions underlying lacunes. Acta Neuropathol 12:1, 1991 98. Okazaki H, Reagan TJ, Campbell RJ. Clinicopathological studies of primary cerebral amyloid angiopathy. Mayo Clin Proc 54:22, 1979 99. Greenberg SM, O'Donnell HC, Schaefer PW, et al: MRI detection of new hemorrhages: potential marker of progression in cerebral
amyloid angiopathy. Neurology 53:1135, 1999 100. Tournier-Lasserve E, Joutel A, Melki J, et al: Cerebral autosomal dominant arteriopathy with subcortical infarcts and
leukoencephalopathy maps to chromosome 19q12. Nat Genet 3:256, 1993 101. Elkind MSV, Sacco RL. Direct thrombin inhibition: a novel approach to stroke prevention in patients
with atrial fibrillation. Stroke 35:1519, 2004 102. Mohr JP, Thompson JL, Lazar RM, et al: Warfarin-Aspirin Recurrent Stroke Study Group. A comparison of warfarin
and aspirin for the prevention of recurrent ischemic stroke. N Engl J Med 345:1444, 2001 103. Algra A, van Gijn J. Aspirin at any dose above 30 mg offers only modest protection after cerebral
ischaemia. J Neurol Neurosurg Psychiatry 60:197, 1996 104. Hass WK, Easton JD, Adams HP Jr, et al: A randomized trial comparing ticlopidine hydrochloride with aspirin for
the prevention of stroke in high-risk patients. Ticlopidine Aspirin Stroke
Study Group. N Engl J Med 321:501, 1989 105. CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients
at risk of ischaemic events (CAPRIE). Lancet 339:1329, 1996 106. Diener HC, Cunha L, Forbes C, et al: European Stroke Prevention Study. 2. Dipyridamole and acetylsalicylic acid
in the secondary prevention of stroke. J Neurol Sci 143:1, 1996 107. Diener HC, for the MATCH Steering Committee. Management of atherothrombosis with clopidogrel in high-risk patients with
recent TIA or ischemic stroke. Presented at the 13th European Stroke Conference, 2004 108. McMahon S, Rodgers A. The epidemiological asscociation between blood pressure and stroke: implications
for primary and secondary prevention. Hypertens Res 17:s23, 1994 109. Wolf PA, Agostino RB, Belanger AJ, et al: Probability of stroke: a risk profile from the Framingham Study. Stroke 22:312, 1991 110. Elkind MS, Sacco RL. Stroke risk factors and stroke prevention. Semin Neurol 18:429, 1998 111. Hansson L, Zanchetti A, Carruthers SG, et al. for the HOT Study Group. Effects of intensive blood pressure lowering and low-dose aspirin in patients
with hypertension: principal results of the Hypertension Optimal
Treatment (HOT) randomised trial. Lancet 351:1755, 1998 112. Chobanian AV, Bakris GL, Black HR, et al: Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and
Treatment of High Blood Pressure. Hypertension 42:1206, 2003 113. MacMahon S, Peto R, Cutler J, et al: Blood pressure, stroke and coronary heart disease: part 1) Prolonged
differences in blood pressure: prospective observational studies corrected
for the regression dilutional bias. Lancet 335:756, 1990 114. Collins R, Peto R, MacMahon S, et al: Blood pressure, stroke and coronary heart disease: part 2) Short term
reductions in blood pressure: overview of randomized trials, their
epidemiological context. Lancet 335:827, 1990 115. Staessen JA, Gasowski J, Wang JG, et al: Risks of untreated and treated isolated systolic hypertension in the elderly: a
meta-analysis of outcome trials. Lancet 355:865, 2000 116. Heart Outcomes Prevention Evaluation (HOPE) Study Investigators. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular
events in high-risk patients. N Engl J Med 342:145, 2000 117. PROGRESS Collaborative Group. Randomised trial of a perindopril-based blood-pressure-lowering regimen
among 6105 individuals with previous stroke or transient ischaemic attack. Lancet 358:1033, 2001 118. The ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research
Group. Major Outcomes in High-Risk Hypertensive Patients Randomized to Angiotensin-Converting
Enzyme Inhibitor or Calcium Channel Blocker vs. Diuretic: the
Antihypertensive and Lipid-Lowering Treatment to Prevent Heart
Attack Trial (ALLHAT). JAMA 288:2981, 2002 119. Iso H, Jacobs DR Jr., Wentworth D, et al: Serum cholesterol levels and six-year mortality from stroke in 350,977 men
screened for the Multiple Risk Factor Intervention Trial. N Eng J Med 320:904, 1989 120. Yano K, Reed DM, MacLean CJ. Serum cholersterol and hemorrhagic stroke in the Honolulu Heart Program. Stroke 20:1460, 1989 121. Hebert PR, Gaziano JM, Chan KS, et al: Cholesterol lowering with statin drugs, risk of stroke, and total mortality: an
overview of randomized trials. JAMA 278:313, 1997 122. Hess DC, Demchuk AM, Brass LM, et al: HMG-CoA reductase inhibitors (statins): a promising approach
to stroke prevention. Neurology 54:790, 2000 123. Albers GW, Hart RG, Lutsep HL, et al: Supplement to the guidelines for the management of transient ischemic attacks. Stroke 30:2502, 1999 124. Sillesen H, for the SPARCL Investigators. Design of the Stroke Prevention by Aggressive Reduction in Cholesterol
Levels (SPARCL) Study. Ongoing Clinical Trials Session, 29th International Stroke Conference, 2004 125. Barrett-Connor E, Khaw K. Diabetes mellitus: an independent risk factor for stroke. Am J Epidemiol 128:116, 1988 126. Bruno A, Biller J, Adams HP, et al: Acute blood glucose level and outcome from ischemic stroke. Trial of ORG 10172 in
Acute Stroke Treatment (TOAST) Investigators. Neurology 52:280, 1999 127. Stratton IM, Adler AI, Neil HAW, et al: Association of glycaemia with macrovascular and microvascular complications
of type 2 diabetes (UKPDS 35): prospective observational
study. BMJ 321:405, 2000 128. Boushey CJ, Beresford AA, Omenn GS, et al: A quantitative assessment of plasma homocysteine as arik factor for vascular
disease. JAMA 274:1049, 1995 129. Fridman O. Hyperhomocysteinemia: atherothrombosis and neurotoxicity. Acta Physiol Ther Latinoam 49:21, 1999 130. Toole JF, Malinow MR, Chambless LE, et al: Lowering homocysteine in patients with ischemic stroke to prevent recurrent
stroke, myocardial infarction, and death: the Vitamin Intervention
for Stroke Prevention (VISP) randomized controlled trial. JAMA 291:565, 2004 131. Hankey GJ, for the VITATOPS Trial Group. The VITATOPS (VITAmins TO Prevent Stroke) Trial: Rationale, Design
and Progress of an International, Large, Simple, Randomised Trial
of Homocysteine-Lowering Multivitamin Therapy in Patients with Recent
Transient Ischaemic Attack or Stroke. 29th International Stroke Conference, 2004 132. Abbott RD, Behrens GR, Sharp DS, et al: Body mass index and thromboembolic stroke in non-smoking men in older middle
age: The Honolulu Heart Program. Stroke 25:2370, 1994 133. Walker SP, Rimm EB, Ascherio A, et al: Body size and fat distribution as predictor of stroke among US men. Am J Epidemiol 144:1143, 1996 134. Kiely DK, Wolf PA, Cupples LA, et al: Physical activity and stroke risk: The Framingham Study. Am J Epidemiol 140:608, 1994 135. Kawachi I, Colditz GA, Stampfer MJ, et al: Smoking cessation and decreased risk of stroke in women. JAMA 269:232, 1993 136. Hillbom M, Kaste M. Ethanol intoxication: a risk factor for ischemic brain infarction in adolescents
and young adults. Stroke 12:422, 1981 137. Klatsky AL. Alcohol and hypertension. Clin Chem Acta 246:91, 1996 138. Thornton JR. Atrial fibrillation in healthy non-alcoholic people after an alcoholic
binge. Lancet 2:1013, 1984 139. Sacco RL, Elkind M, Boden-Albala B, et al: The protective effect of moderate alcohol drinking on ischemic stroke. JAMA 281:53, 1999 140. Berger K, Ajani UA, Kase C, et al: Light-to-moderate alcohol consumption and the risk of stroke among US male
physicians. N Engl J Med 341:1557, 1999 141. Finucane FF, Madans JH, Bush TL, et al: Decreased risk of stroke among postmenopausal hormone users: results from
a national cohort. Arch Intern Med 153:73, 1993 142. Writing Group for the Women's Health Initiative Investigators. Risks and Benefits of Estrogen Plus Progestin in Healthy Postmenopausal
Women: Principal Results From the Women's Health Initiative Randomized
Controlled Trial. JAMA 288:321, 2002 143. The Women's Health Initiative Steering Committee. Effects of Conjugated Equine Estrogen in Postmenopausal Women With Hysterectomy: The
Women's Health Initiative Randomized Controlled Trial. JAMA 291:1701, 2004 144. Viscoli CM, Brass LM, Kernan WN, et al: A clinical trial of estrogen-replacement therapy after ischemic stroke. N Engl J Med 345:1243, 2001 145. Hulley S, Gradt D, Bush T, et al: Randomized trial of estrogen plus progestin for secondary prevention of
coronary heart disease in postmenopausal women. JAMA 280:605, 1998 |