NORMAL FUNCTION: THE VESTIBULAR REFLEXES
Vestibular reflexes are triggered by head movements. Imagine a bird watcher standing in a rocking boat traveling downstream (Fig. 1A). To identify a bird roosting on a tree, the image of the bird must be kept stable on the retina by three separate vestibular reflexes (see Fig. 1B). As the boat pitches up and down the eyes must move in the opposite direction and synchronously with the angular motion of the head to keep the eyes stable in space. This is accomplished by the angular vestibulo-ocular reflex (VOR), which is sensed by the semicircular canals in the inner ear (see Fig. 1B). As the boat moves down the river toward the bird, the eyes must move horizontally in the opposite direction synchronously with the linear motion of the boat. This is accomplished by the linear VOR, which is sensed by the otoliths in the inner ear (see Fig. 1C). Figure 1D shows the most complicated reflex as the boat tilts left and right. To keep the head and eyes level during tilt to the right, the right eye is elevated in the orbit and the left eye is depressed (skew deviation). Both eyes undergo torsion to the left within the orbit (counter-roll deviation) and the head is tilted to the left on the body (head tilt). This is accomplished by the ocular tilt reflex, which is sensed by the otoliths. Without these vestibular reflexes, visual acuity would be degraded and diplopia would occur. Table 1 lists the three vestibular reflexes, the sensory organs involved, and the motor output for each reflex.
|
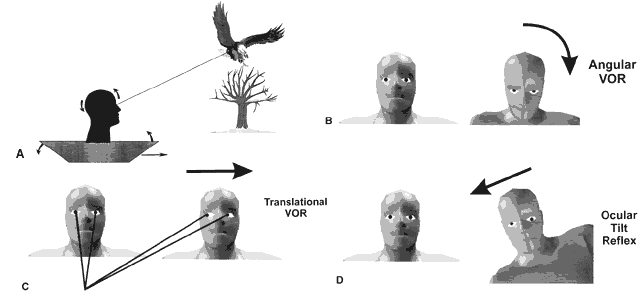
TABLE 18-1. Vestibular Reflexes
| Vestibular Reflex | Sensory Organ | Motor Output |
| Angular VOR | Semicircular canals (SCCs) Horizontal Posterior Anterior | Eyes move opposite to angular movement (rotation) of the head. Shaking the head up and down (yes) is termed pitch and is sensed by the anterior and posterior SCCs. Shaking the head horizontally (no) is termed yaw and is sensed by the horizontal SCCs. |
| Linear VOR | Otoliths Saccule Utricle | Eyes move opposite to linear movement of the head. Linear movement up and down (riding in an elevator) is sensed by the saccule. Linear movement horizontally (riding in a train on a straight track) is sensed by the utricle. |
| Ocular tilt reflex | Otolith Utricle | Eyes and head move opposite to tilt of the head. Tilt left causes elevation of the left eye, depression of the right eye, torsion of both eyes to the right, and rightward tilt of the head. |
VOR, vestibulo-ocular reflex.
PATHOPHYSIOLOGY
Peripheral Structures
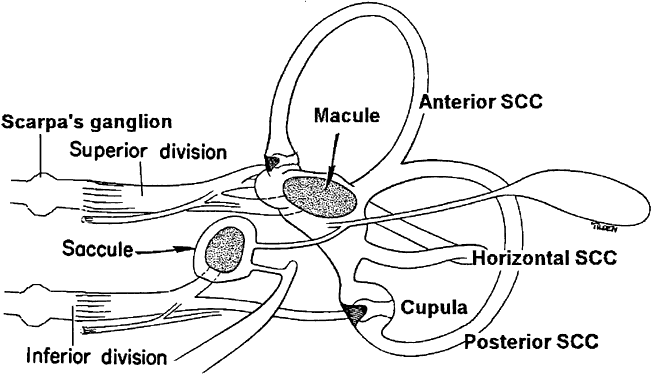
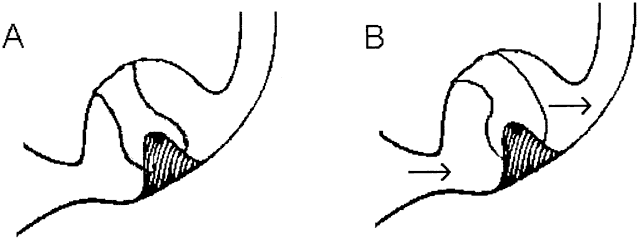
The vestibular sensory organs lie within the membranous labyrinth of the inner ear, protected in the petrous portion of the temporal bone. The membranous labyrinth consists of three semicircular canals (SCCs)—the anterior, posterior, and horizontal—that lie 90 degrees (orthogonal) to each other, and two otoliths, the utricle (horizontally aligned) and the saccule (vertically aligned) (Fig. 2). The labyrinth is innervated by the vestibular nerve, which is part of the VIIIth nerve. The vestibular nerve contains two fascicles, the superior and inferior divisions. The cell bodies of each axon of the VIIIth nerve lie in Scarpa's ganglion, located in the internal auditory canal. The superior division innervates the utricle and the anterior and horizontal SCCs. The inferior division innervates the saccule and posterior SCCs. Within each semicircular canal is an area of hair cells that protrude theirprocesses into a gelatinous matrix called the cupula. Angular head acceleration imposes inertial forces on the endolymph fluid within the canal, which causes relative fluid flow through the canal in the direction opposite to that of head acceleration. This flow deflects the cupula and bends the hair cells (Fig. 3). During head acceleration, these hair cells bend in proportion to head acceleration and change the neuronal firing rate in the VIIIth nerve. Because each Scarpa's ganglion is spontaneously “firing” at 100 spike/sec, head motion (acceleration or deceleration) modulates this firing rate. The firing rate is increased for ipsilateral angular head movements and decreased for contralateral angular head movements. The otoliths also contain a local region of hair cells. The hair cells protrude their processes into a gelatinous matrix called the macula, which is covered by a surface of calcium carbonate crystals, the otoconia. The otolith organs respond to linear acceleration and sustained head tilt relative to gravity. Linear acceleration (including tilt of the head) causes these crystals to move, which bends the hair cells and modulates the firing rate in the VIIIth nerve. The firing rate is increased for ipsilateral linear head movement or tilt, and is decreased for contralateral linear head movement and tilt.
|
|
Semicircular Canals
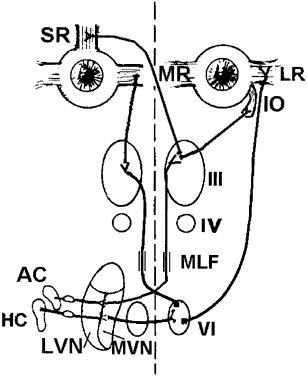
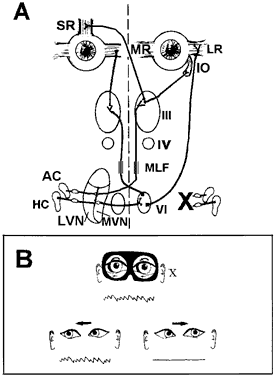
Each SCC innervates two eye muscles by means of a three-neuron arc. The central connections of the horizontal and anterior SCCs on one side are shown in Figure 4. The projections of these two SCCs are shown together because usually they are both involved in vestibular neuritis, the most common cause of severe vertigo. The pathophysiology of vestibular neuritis is diagrammed in Figure 5. This disorder disrupts the superior division of the vestibular nerve.2 Because the Scarpa's ganglion on each side normally is firing at 100 spikes/sec, any loss of activity on one side results in relative excessive excitation from the intact side. This large bias in neural activity causes nystagmus. The direction of nystagmus is determined according to the quick phase, but the vestibular deficit is actually driving the slow phase of the nystagmus. Vestibular neuritis results in a mixed horizontal and torsional nystagmus. This pattern of nystagmus is caused by the innervation pattern of the superior division of the VIIIth nerve on the intact side (recall that the superior division innervates the horizontal and anterior semicircular canals; see Fig. 2). Relative excitation of the horizontal SCC causes horizontal nystagmus with the slow phase toward the side of the lesion. Figure 5 depicts a left-sided lesion, which would cause a right-beating nystagmus. Relative excitation of the anterior SCC causes torsional nystagmus (counterclockwise nystagmus for a left-sided lesion). Because of the confusion over whether to label torsional nystagmus from the perspective of the observer or the patient, the current trend is to assess torsional nystagmus according to the direction of the quick phases toward which the superior poles of both eyes are beating. Thus, a left-sided lesion results in right beating and right torsional nystagmus (see Fig. 5B). There are two other key features of vestibular nystagmus. The intensity of nystagmus is increased when fixation is blocked (Fig. 5B depicts increased nystagmus when the subject looks through Frenzel or 20-diopter lenses), and the intensity of nystagmus also increases when the patient looks in the direction of the quick phases.
|
|
Otoliths
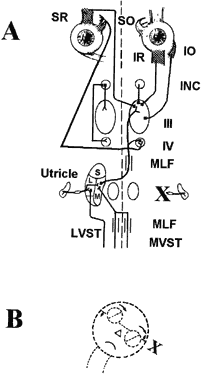
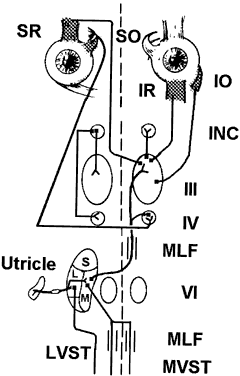
Each otolith innervates four eye muscles by means of a three-neuron arc. The central connections of the utricle on one side are shown in Figure 6.3 The projection to the vertical muscles causes vertical eye deviation and torsion during head tilt. The projections in the lateral and medial vestibulospinal tracts mediate the head tilt during the ocular tilt reflex. Acute loss of function of the utricle on one side from the VIIIth nerve section or vestibular neuritis causes a pathologic ocular tilt response because of the unopposed excitation of the intact utricle. Figure 7 depicts the results of a left-sided lesion.4 Excitation of the right superior rectus and oblique muscles causes elevation and intorsion of that eye. Excitation of the left inferior rectus and oblique muscles causes depression and extorsion of that eye. Excitation of the neck muscles innervated by the intact vestibulospinal tracts causes a left head tilt.
|
|