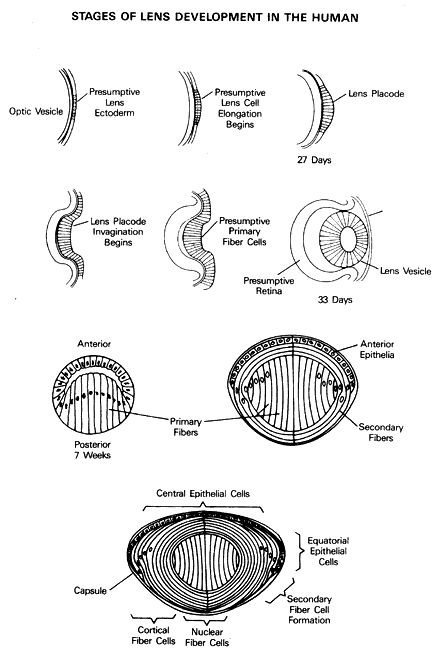
1. Francois J: Genetics of cataract. Ophthalmologica 184:61, 1982 2. Lerman S: Radiant Energy and the Eye. New York: Macmillan, 1980 3. Benedek GB: Theory of transparency of the eye. Appl Optics 10:459, 1971 4. Delaye M, Tardieu A: Short-range order of crystallin proteins accounts for eye lens transparency. Nature 302:415, 1983 5. Bettelheim FA, Siew EL: Effect of changes in concentration upon lens turbidity as predicted by
the random fluctuation theory. Biophys J 41:29, 1983 6. Delaye M, Gromiec A: Mutual diffusion of crystallin proteins at finite concentrations: A light
scattering study. Biopolymers 22:1203, 1983 7. Vrensen G, Kappelhof J,, Willikens B: Aging of the human lens. Lens Eye Tox Res 7:1, 1990 8. Kuszak JR, Deutsch TA,, Brown HG: Anatomy of aged, senile cataractous lenses. In
Albert D, Jacobiec F (eds): Principles, Practice of Ophthalmology: Basic
Sciences, p 82. Philadelphia: WB Saunders, 1994 9. Harding CV, Maisel H,, Chylack LT: The structure of the human cataractous
lens. In Maisel H (ed): The Ocular Lens, p 405. New York: Marcel Dekker
Inc., 1985 10. Benedek GB, Chylack LT, Libondi T et al: Quantitative detection of the molecular changes associated with early cararactogenesis
in the living human lens using quasielastic light scattering. Curr Eye Res 6:1421, 1987 11. Bettelheim FA: Physical basis of lens transparency. In Maisel H (ed): The
Ocular Lens, p 265. New York: Marcel Dekker Inc, 1985 12. Benedek GB, Clark JI, Serrallach EU et al: Light scattering and reversible cataracts in calf and human lens. Phil Trans R Soc Lond A 293:329, 1979 13. Merin S, Crawford JS: The etiology of congenital cataracts. A survey of 386 cases. Can J Ophthal 6:178, 1971 14. Semina EV, Ferrell RE, Mintz-Hittner HA et al: A novel homeobox gene PITX3 is mutated in families with autosomal-dominant
cataracts, ASMD. Nat Genet 19:167, 1998 15. Hejtmancik JF: The genetics of cataract: Our vision becomes clearer. Am J Hum Genet 62:520, 1998 16. Ehling UH: Genetic risk assessment. Annu Rev Genet 25: 255, 1991 17. Harding JJ, Rixon KC, Marriott FHC: Men have heavier lenses than women of the same age. Exp Eye Res 25: 651, 1977 18. Wistow GJ, Piatigorsky J: Lens crystallins: The evolution and expression of proteins for a highly
specialized tissue. Annu Rev Biochem 57:479, 1988 19. Mann I: The Development of the Human Eye. New York: Grune, Stratton, 1964 20. Kuzak JR: Embryology and anatomy of the lens. In Tasman W, Jaeger EA (eds): Duane's
Clinical Ophthalmology, p 1. Philadelphia: JB Lippincott, 1990 21. Halder G, Callaerts P, Gehring WJ: Induction of ectopic eyes by targeted expression of the eyeless gene in
Drosophila. Science 267:1788, 1995 22. Mathers PH, Grinberg A, Mahon KA, Jamrich M: The Rx homeobox gene is essential for vertebrate eye development. Nature 387:603, 1997 23. Hanson I, van Heyningen V: Pax-6: More than meets the eye. Trends Genet 11:268, 1995 24. Walther C, Gruss P: Pax-6, a murine paired box gene, is expressed in the developing CNS. Development 113: 1435, 1991 25. Fujiwara M, Uchida T, Osumi-Yamashita N, Eto K: Uchida rat (rSey): A new mutant rat with craniofacial abnormalities resembling
those of the mouse Sey mutant. Differentiation 57:31, 1994 26. Brewer C, Holloway S, Zawalnyski P et al: A chromosomal deletion map of human malformations. Am J Hum Genet 63:1153, 1998 27. Oliver G, Loosli F, Koster R et al: Ectopic lens induction in fish in response to the murine homeobox gene
Six3. Mech Dev 60:233, 1996 28. Nishiguchi S, Wood H, Kondoh H et al: Sox1 directly regulates the gamma-crystallin genes and is essential for
lens development in mice. Genes Dev 12:776, 1998 29. Semina EV, Reiter RS, Murray JC: Isolation of a new homeobox gene belonging to the PITX/RIEG family: Expression
during lens development and mapping to the aphakia region on mouse
chromosome 19. Hum Mol Genet 6:2109, 1997 30. Rafferty NS: Lens morphology. In Maisel H (ed): The Ocular Lens, p 1. New
York: Marcel Dekker Inc., 1985 31. Goodenough DA, Dick JSB, Lyons JE: Lens metabolic cooperation: A study of mouse lens transport, permeability
visualized with freeze-substitution autoradiography, electron microscopy. J Cell Biol 86:576, 1980 32. Gorthy WC, Snavely MR, Berrong ND: Some aspects of transport and digestion in the lens of the normal young
adult rat. Exp Eye Res 12:112, 1971 33. Rae JL, Stacey T: Lanthanum and procion yellow as extracellular markers in the crystalline
lens of the rat. Exp Eye Res 28:1, 1979 34. Ramaekers FCS, Bloemendal H: Cytoskeletal, contractile structures in lens
cell differentiation. In Bloemendal H (ed): Molecular and Cellular
Biology of the Eye Lens, p 85. New York: John Wiley & Sons, 1981 35. Benedetti L, Dunia I, Ramaekers FCS, Kibbelaar MA: Lenticular plasma membranes
and cytoskeleton. In Bloemendal H (ed): Molecular and Cellular
Biology of the Eye Lens, p 137. New York: John Wiley & Sons, 1981 36. Alcala H, Maisel H: Biochemistry of lens plasma membranes and cytoskeleton. In
Maisel H (ed): The Ocular Lens, p 169. New York: Marcel Dekker
Inc., 1985 37. Ireland M, Maisel H: A family of lens fiber cell specific proteins. Lens Eye Tox Res 6:623, 1989 38. Piatigorsky J: Gene expression and genetic engineering in the lens. Invest Ophthalmol Vis Sci 28:9, 1987 39. Ostrer H, Beebe DC, Piatigorsky J: Beta-crystallin mRNAs: Differential distribution in the developing chicken
lens. Dev Biol 86:403, 1981 40. Hejtmancik JF, Beebe DC, Ostrer H, Piatigorsky J: Delta-, beta-crystallin mRNA levels in the embryonic, posthatched chicken
lens: Temporal, spatial changes during development. Dev Biol 109:72, 1985 41. Van Leen RW, Van Roozendaal KEP, Lubsen NH, Schoenmakers JG: Differential expression of crystallin genes during development of the rat
eye lens. Dev Biol 120:457, 1987 42. Aarts HJ, Lubsen NH, Schoenmakers JG: Crystallin gene expression during rat lens development. Eur J Biochem 183:31, 1989 43. Merin S: Congenital cataracts. In Goldberg MF (ed): Genetic, Metabolic
Eye Disease, p 337. Boston: Little, Brown & Co, 1974 44. Al-Ghoul KJ, Novak LA, Kuszak JR: The structure of posterior subcapsular cataracts in the Royal College of
Surgeons (RCS) rats. Exp Eye Res 67:163, 1998 45. Berry V, Ionides AC, Moore AT et al: A locus for autosomal dominant anterior polar cataract on chromosome 17p. Hum Mol Genet 5:415, 1996 46. Ionides AC, Berry V, Mackay DS et al: A locus for autosomal dominant posterior polar cataract on chromosome 1p. Hum Mol Genet 6:47, 1997 47. Eiberg H, Lund AM, Warburg M, Rosenberg T: Assignment of congenital cataract Volkmann type (CCV) to chromosome 1p36. Hum Genet 96:33, 1995 48. Renwick JH, Lawler SD: Probable linkage between a congenital cataract locus and the Duffy blood
group locus. Ann Hum Genet 27:67, 1963 49. Lubsen NH, Renwick JH, Tsui LC et al: A locus for a human hereditary cataract is closely linked to the gamma-crystallin
gene family. Proc Natl Acad Sci USA 84:489, 1987 50. Mackay D, Ionides A, Berry V et al: A new locus for dominant “zonular pulverulent” cataract, on
chromosome 13. Am J Hum Genet 60:1474, 1997 51. Richards J, Maumenee IH, Rowe S, Lourien EW: Congenital cataract possibly linked to haptoglobin. Cytogenet Cell Genet 37:570, 1984 52. Basti S, Hejtmancik JF, Padma T et al: Autosomal dominant zonular cataract with sutural opacities in a four-generation
family. Am J Ophthalmol 121:162, 1996 53. Kuszak JR: Embryology and anatomy of the lens. In Tasman W, Jaeger EA (eds): Duane's
Clinical Ophthalmology. Philadelphia: JB Lippincott, 1990 54. Kaiser-Kupfer MI, Kuwabara T, Uga S et al: Cataract in gyrate atrophy—clinical and morphological studies. Invest Ophthalmol Vis Sci 24:432, 1983 55. Morner CT: Untersuchungender protein-substanzen in den lichtbrechenden Medien des
Auges. Hoppe-Seyler's Z Physiol Chem 18:61, 1894 56. de Jong WW, Terwindt EC, Bloemendal H: The amino acid sequence of the A chain of human alpha-crystallin. FEBS Lett 58:310, 1975 57. Kramps JA, deMan BM, de Jong WW: The primary structure of the B2 chain of human alpha-crystallin. FEBS Lett 74:82, 1977 58. van der Ouderaa FJ, de Jong WW, Hilderink A, Bloemendal H: The amino acid sequence of the alphaB2 chain of bovine alpha-crystallin. Eur J Biochem 49:157, 1974 59. Horwitz J: Some properties of the low-molecular-weight alpha-crystallin from normal
human lens: Comparison with bovine lens. Exp Eye Res 19:49, 1974 60. Tardieu A, Laporte D, Licinio P et al: Calf lens alpha-crystallin quaternary structure. A three-layer model. J Mol Biol 192:711, 1986 61. Augusteyn RC, Koretz JF: A possible structure for alpha-crystallin. FEBS Lett 222:1, 1987 62. Thomson JA, Augusteyn RC: On the structure of alpha-crystallin: Construction of hybrid molecules, homopolymers. Biochim Biophys Acta 994:246, 1989 63. Hendriks W, Weetink H, Voorter CE et al: The alternative splicing product alpha Ains-crystallin is structurally
equivalent to alpha A, alpha B subunits in the rat alpha-crystallin aggregate. Biochim Biophys Acta 1037:58, 1990 64. Bova MP, Ding LL, Horwitz J, Fung BK: Subunit exchange of alpha A-crystallin. J Biol Chem 272:29511, 1997 65. Sun TX, Liang JN: Intermolecular exchange, stabilization of recombinant human alpha A- and
alpha B-crystallin. J Biol Chem 273:286, 1998 66. Haley DA, Horwitz J, Stewart PL: The small heat-shock protein, alpha B-crystallin, has a variable quaternary
structure. J Mol Biol 277:27, 1998 67. Brady JP, Garland D, Duglas-Tabor Y et al: Targeted disruption of the mouse alpha A-crystallin gene induces cataract
and cytoplasmic inclusion bodies containing the small heat shock protein
alpha B-crystallin. Proc Natl Acad Sci USA 94:884, 1997 68. Wawrousek EF, Brady JP: Alpha B-crystallin gene knockout mice develop a severe, fatal phenotype
late in life. Invest Ophthalmol Vis Sci 39:S523, 1998 69. Kantorow M, Piatigorsky J: Alpha-crystallin/small heat shock protein has autokinase activity. Proc Natl Acad Sci USA 91:3112, 1994 70. Kantorow M, Horwitz J, van Boekel MA et al: Conversion from oligomers to tetramers enhances autophosphorylation by
lens alpha A-crystallin. Specificity between alpha A- and alpha B-crystallin
subunits. J Biol Chem 270:17215, 1995 71. Sun TX, Das BK, Liang JJ: Conformational and functional differences between recombinant human lens
alpha A- and alpha B-crystallin. J Biol Chem 272:6220, 1997 72. Horwitz J, Huang QL, Ding L, Bova MP: Lens alphacrystallin: Chaperone-like properties. Methods Enzymol 290:365, 1998 73. Horwitz J: Alpha-crystallin can function as a molecular chaperone. Proc Natl Acad Sci USA 89:10449, 1992 74. Muchowski PJ, Bassuk JA, Lubsen NH, Clark JI: Human alpha B-crystallin. Small heat shock protein and molecular chaperone. J Biol Chem 272:2578, 1997 75. Raman B, Rao CM: Chaperone-like activity and temperature-induced structural changes of alpha-crystallin. J Biol Chem 272:23559, 1997 76. Bhat SP, Horwitz J, Srinivasan A, Ding L: AlphaB-crystallin exists as an independent protein in the heart and in
the lens. Eur J Biochem 202:775, 1991 77. Litt M, Kramer P, LaMorticella DM et al: Autosomal dominant congenital cataract associated with a missense mutation
in the human alpha crystallin gene CRYAA. Hum Mol Genet 7:471, 1998 77a. Bova MP, Yaron O, Huang Q et al: Mutation R120G in alphaB-crystallin, which is linked to a desmin-related
myopathy, results in an irregular structure and defective chaperone-like
function. Proc Natl Acad Sci USA 96:6137, 1999 78. Vicart P, Caron A, Guicheney P et al: A missense mutation in the αB-crystallin chaperone gene causes a desmin-related
myopathy. Nat Genet 20:92, 1998 79. Driessen HP, Herbrink P, Bloemendal H, de Jong WW: Primary structure of the bovine beta-crystallin Bp chain. Internal duplication
and homology with gamma-crystallin. Eur J Biochem 121:83, 1981 80. Wistow G, Slingsby C, Blundell T et al: Eye-lens proteins: The three-dimensional structure of beta-crystallin predicted
from monomeric gamma-crystallin. FEBS Lett 133:9, 1981 81. Lubsen NH, Aarts HJM, Schoenmakers JGG: The evolution of lenticular proteins: The beta- and gamma-crystallin supergene
family. Prog Biophys Mol Biol 51:47, 1988 82. Blundell T, Lindley P, Miller L et al: The molecular structure and stability of the eye lens: X-ray analysis of
gamma-crystallin II. Nature 289:771, 1981 83. Wistow GJ, Turnell B, Summers L et al: X-ray analysis of the eye lens protein gamma-II crystallin at 1.9 A resolution. J Mol Biol 170:175, 1983 84. White HE, Driessen HPC, Slingsby C et al: Packing interactions in the eye-lens. Structural analysis, internal symmetry, lattice
interactions of bovine gamma IVa-crystallin. J Mol Biol 207:217, 1989 85. Norledge BV, Hay RE, Bateman OA et al: Towards a molecular understanding of phase separation in the lens: A comparison
of the X-ray structures of two high Tc gamma-crystallins, gammaE
and gammaF, with two low Tc gamma-crystallins, gammaB and gammaD. Exp Eye Res 65:609, 1997 86. Siezen RJ, Wu E, Kaplan ED et al: Rat lens gamma-crystallins. Characterization of the six gene products and
their spatial and temporal distribution resulting from differential
synthesis. J Mol Biol 199:475, 1988 87. Broide ML, Berland CR, Pande J et al: Binary-liquid phase separation of lens protein solutions. Proc Natl Acad Sci USA 88:5660, 1991 88. Tanaka T, Benedek GB: Observation of protein diffusivity in intact human and bovine lenses with
application to cataract. Invest Ophthalmol Vis Sci 14:449, 1975 89. Wistow G: Evolution of a protein superfamily: Relationships between vertebrate lens
crystallins and microorganism dormancy proteins. J Mol Evol 30:140, 1990 90. Krasko A, Muller IM, Muller WE: Evolutionary relationships of the metazoan beta gamma-crystallins, including
that from the marine sponge Geodia cydonium. Proc R Soc Lond B Biol Sci 264:1077, 1997 91. Ray ME, Wistow G, Su YA et al: A1M1, a novel non-lens member of the beta-gamma-crystallin superfamily
associated with the control of tumorigenicity in human malignant melanoma. Proc Natl Acad Sci USA 94:3229, 1997 92. van Dam AF: Purification and composition studies of betaS-crystallin. Exp Eye Res 5:255, 1966 93. Quax-Jeuken Y, Driessen H, Leunissen J et al: BetaScrystallin: Structure and evolution of a distinct member of the beta
gamma-superfamily. EMBO J 4:2597, 1985 94. van Rens GL, Raats JM, Driessen HP et al: Structure of the bovine eye lens gamma s-crystallin gene (formerly beta
s). Gene 78:225, 1989 95. Zigler JS Jr, Russell P, Horwitz J et al: Further studies on low-molecular-weight crystallins: Relationship between
the bovine beta s, the human 24 kD protein and the gamma-crystallins. Curr Eye Res 5:395, 1986 96. den Dunnen JT, Jongbloed RJE, Geurts van Kessel AHM, Schoenmakers JGG: Human lens gamma-crystallin sequences are located in the p12-qter region
of chromosome 2. Hum Genet 70:217, 1985 97. Shiloh Y, Donlon T, Bruns G et al: Assignment of the human gamma-crystallin gene cluster (CRYG) to the long
arm of chromosome 2, region q33-36. Hum Genet 73:17, 1986 98. Slingsby C, Croft LR: Developmental changes in the low-molecular-weight proteins of the bovine
lens. Exp Eye Res 17:369, 1973 99. McDevitt DS, Croft LR: On the existence of gammacrystallin in the bird lens. Exp Eye Res 25:473, 1977 100. van Rens GL, de Jong WW, Bloemendal H: One member of the gamma-crystallin gene family, gamma s, is expressed in
birds [letter]. Exp Eye Res 53:135, 1991 101. Heon E, Priston M, Schorderet DF et al: The gamma-crystallins and human cataracts: A puzzle made clearer. Am J Hum Genet 65:1261, 1999 102. Ren Z, Li A, Shastry BS et al: A 5-base insertion in the C-crystallin gene
is associated with autosomal dominant variable zonular pulverulent
cataract. Hum Genet, DOI 10.1007/s004390000289 103. Kmoch S, Asfaw B, Bezouska K et al: Cataract due to crystal deposits of 37R→S mutated gammaD-crystallin. Am J Hum Genet 65:A305, 1999 104. Peterson CA, Piatigorsky J: Preferential conservation of the globular domains of the beta A3/A1-crystallin
polypeptide of the chicken eye lens. Gene 45:139, 1986 105. Hejtmancik JF, Thompson MA, Wistow G, Piatigorsky J: cDNA and deduced protein sequence for the beta B1crystallin polypeptide
of the chicken lens: Conservation of the PAPA sequence. J Biol Chem 261:982, 1986 106. Berbers GAM, Hoekman WA, Bloemendal H et al: Homology between the primary structures of the major bovine beta-crystallin
chains. Eur J Biochem 139:467, 1984 107. Berbers GAM, Hoekman WA, Bloemendal H et al: Proline- and alanine-rich N-terminal extension of the basic bovine beta-crystallin
B1 chains. FEBS Lett 161:225, 1983 108. Kleiman NJ, Chiesa R, Kolks MA, Spector A: Phosphorylation of beta-crystallin B2 (beta Bp) in the bovine lens. J Biol Chem 263:14978, 1988 109. Voorter CE, Bloemendal H, de Jong WW: In vitro and in vivo phosphorylation of chicken beta B3-crystallin. Curr Eye Res 8:459, 1989 110. Wistow G, Roquemore E, Kim HS: Anomalous behavior of betaB1-crystallin subunits from avian lenses. Curr Eye Res 10:313, 1991 111. Head MW, Peter A, Clayton RM: Evidence for the extralenticular expression of members of the beta-crystallin
gene family in the chick and a comparison with delta-crystallin
during differentiation and transdifferentiation. Differentiation 48:147, 1991 112. Head MW, Sedowofia K, Clayton RM: Beta B2-crystallin in the mammalian retina. Exp Eye Res 61:423, 1995 113. Dirks RP, van GS, Kruse JJ et al: Extralenticular expression of the rodent beta B2-crystallin gene. Exp Eye Res 66: 267, 1998 114. Kantorow M, Horwitz J, Sergeev YV et al: Extralenticular expression cAMP-dependent kinase phosphorylation and autophosphorylation
of beta B2-crystallin. Invest Ophthalmol Vis Sci 38:S205, 1997 115. Smolich BD, Tarkington SK, Saha MS, Grainger RM: Xenopus gamma-crystallin gene expression: Evidence that the gamma-crystallin gene
family is transcribed in lens and nonlens tissues. Mol Cell Biol 14:1355, 1994 116. Brunekreef GA, van GS, Destree OH, Lubsen NH: Extralenticular expression of Xenopus laevis alpha-, beta-, and gamma-crystallin genes. Invest Ophthalmol Vis Sci 38: 2764, 1997 117. Sinha D, Esumi N, Jaworski C et al: Cloning and mapping the mouse Crygs gene and non-lens expression of gamma
S-crystallin. Mol Vis 4:8, 1998 118. Heon E, Munier F, Tsilfidis C, Liu S: Mapping of congenital aculeiform cataract to chromosome 2q33. Invest Ophthalmol Vis Sci 38:S934, 1997 119. Padma T, Ayyagari R, Murty JS et al: Autosomal dominant zonular cataract with sutural opacities localized to
chromosome 17q11-12. Am J Hum Genet 57:840, 1995 120. Kannabiran C, Rogan PK, Olmos L et al: Autosomal dominant zonular cataract
with sutural opacities is associated with a splice site mutation in
the αA3/A1-crystallin gene. Mol Vis 4:1998 121. Geyer DD, Johannes M, Masser DS et al: Identification of a novel locus for autosomal dominant congenital cataract
on chromosome 17q. Invest Ophthalmol Vis Sci 39:S419, 1998 122. Litt M, Carrero-Valenzuela R, LaMorticella DM et al: Autosomal dominant cerulean cataract is associated with a chain termination
mutation in the human beta-crystallin gene CRYBB2. Hum Mol Genet 6:665, 1997 123. de Jong WW, Hendriks W, Mulders JW, Bloemendal H: Evolution of eye lens crystallins: The stress connection. Trends Biochem Sci 14:365, 1989 124. Piatigorsky J, Wistow GJ: Enzyme/crystallins: Gene sharing as an evolutionary strategy. Cell 57:197, 1989 125. Piatigorsky J, Wistow GJ: The recruitment of crystallins: New functions precede gene duplication. Science 252: 1078, 1991 126. Piatigorsky J: Gene sharing in lens and cornea: Facts and implications. Prog Ret Eye Res 17:145, 1998 127. Atreya PL, Barnes J, Katar M et al: N-cadherin of the human lens. Curr Eye Res 8:947, 1989 128. Watanabe M, Kobayashi H, Rutishauser U et al: NCAM in the differentiation of embryonic lens tissue. Dev Biol 135:414, 1989 129. Xu KY, Becker LC: Ultrastructural localization of glycolytic enzymes on sarcoplasmic reticulum
vesicles. J Histochem Cytochem 46:419, 1998 130. Mulders JW, Voorter CE, Lamers C et al: MP17, a fiber-specific intrinsic membrane protein from mammalian eye lens. Curr Eye Res 7:207, 1988 131. Chepelinsky AB: The MIP transmembrane channel gene family. In Peracchia
C (ed): Handbook of Membrane Channels: Molecular and Cellular Physiology, p 413. New
York: Academic Press, 1994 132. Park JH, Saier MH Jr: Phylogenetic characterization of the MIP family of transmembrane channel
proteins. J Membr Biol 153:171, 1996 133. Yang B, Verkman AS: Water and glycerol permeabilities of aquaporins 1–5 and MIP determined
quantitatively by expression of epitope-tagged constructs in Xenopus oocytes. J Biol Chem 272:16140, 1997 134. Cheng A, van HA, Yeager M et al: Three-dimensional organization of a human water channel. Nature 387:627, 1997 135. Saito F, Sasaki S, Chepelinsky AB et al: Human AQP2 and MIP genes, two members of the MIP family, map within chromosome
band 12q13 on the basis of two-color FISH. Cytogenet Cell Genet 68:45, 1995 136. Gorin MB, Yancey SB, Cline J et al: The major intrinsic protein (MIP) of the bovine lens fiber membrane: Characterization, structure
based on cDNA cloning. Cell 39:49, 1984 137. Schey KL, Fowler JG, Schwartz JC et al: Complete map and identification of the phosphorylation site of bovine lens
major intrinsic protein. Invest Ophthalmol Vis Sci 38: 2508, 1997 138. Zampighi GA, Gall JE, Ehring GR, Simon SA: The structural organization and protein compositions of lens fiber junctions. J Cell Biol 108:2255, 1989 139. FitzGerald PG, Bok D, Horwitz J: Immunocytochemical localization of the main intrinsic polypeptide (MIP) in
ultrathin frozen sections of rat lens. J Cell Biol 97:1491, 1983 140. Gooden MM, Rintoul DA, Takehana M, Takemoto L: Major intrinsic polypeptide (MIP 26K) from lens membrane: Reconstitution
into vesicles and inhibition of channel forming activity by peptide
antiserum. Biochem Biophys Res Commun 128:993, 1985 141. Peracchia C, Girsch SJ: Permeability and gating of lens gap junction channels incorporated into
liposomes. Curr Eye Res 4:431, 1985 142. Zampighi GA, Hall JE, Kreman M: Purified lens junctional protein forms channels in planner lipid films. Proc Natl Acad Sci USA 82:8468, 1985 143. Ehring GR, Zampighi G, Horwitz J et al: Properties of channels reconstituted from the major intrinsic protein of
lens membranes. J Gen Physiol 96:631, 1990 144. Louis CF, Hogan P, Visco L, Strasburg G: Identity of the calmodulin-binding proteins in bovine lens plasma membranes. Exp Eye Res 50:495, 1990 145. Garland D, Russell P: Phosphorylation of lens fiber cell membrane proteins. Proc Natl Acad Sci USA 82:653, 1985 146. Manenti S, Dunia I, Benedetti EL: Fatty acid acylation of lens fiber plasma membrane proteins. MP26 and alpha-crystallin
are palmitoylated. FEBS Lett 262:356, 1990 147. Geyer DD, Le X, Johannes M et al: DNA sequence studies of the major intrinsic protein (MIP) gene in an autosomal
dominant cataract family (ADC2). Invest Ophthalmol Vis Sci 40:S786, 1999 148. Jiang JX, Goodenough DA: Heteromeric connexins in lens gap junction channels. Proc Natl Acad Sci USA 93:1287, 1996 149. Kistler J, Christie D, Bullivant S: Homologies between gap junction proteins in lens, heart and liver. Nature 331:721, 1988 150. Gruijters WT, Kistler J, Bullivant S, Goodenough DA: Immunolocalization of MP70 in lens fiber 16–17-nm intercellular junctions. J Cell Biol 104:565, 1987 151. Kistler J, Schaller J, Sigrist H: MP38 contains the membrane-embedded domain of the lens fiber gap junction
protein MP70. J Biol Chem 265:13357, 1990 152. Arneson ML, Cheng HL, Louis CF: Characterization of the ovine-lens plasma-membrane protein-kinase substrates. Eur J Biochem 234:670, 1995 153. Berthoud VM, Beyer EC, Kurata WE et al: The gapjunction protein connexin 56 is phosphorylated in the intracellular
loop and the carboxy-terminal region. Eur J Biochem 244:89, 1997 154. Church RL, Wang JH, Steele E: The human lens intrinsic membrane protein MP70 (Cx50) gene: Clonal analysis
and chromosome mapping [published erratum appears in Curr Eye
Res 14:979, 1995]. Curr Eye Res 14:215, 1995 155. Geyer DD, Church RL, Steele ECJ et al: Regional mapping of the human MP70 (Cx50; connexin 50) gene by fluorescence
in situ hybridization to 1q21 1. Mol Vis 3:13, 1997 156. Nettleship E, Ogilvie FM: A peculiar form of hereditary congenital cataract. Trans Ophthal Soc UK 26:191, 1906 157. Shiels A, Mackay D, Ionides A et al: A missense mutation in the human connexin 50 gene (GJA8) underlies autosomal
dominant “zonular pulverulent” cataract, on chromosome 1q. Am J Hum Genet 62:526, 1998 158. Mackay D, Ionides A, Kibar Z et al: Connexin46 mutations in autosomal dominant congenital cataract. Am J Hum Genet 64:1357, 1999 159. Gong X, Li E, Klier G, Huang Q et al: Disruption of alpha3-connexin gene leads to proteolysis and cataractogenesis
in mice. Cell 91:833, 1997 160. White TW, Goodenough DA: Ocular abnormalities in connexin 50 knockout mice. Mol Biol Cell 8:93, 1997 161. Allen DP, Low PS, Dola A, Maisel H: Band 3 and ankyrin homologues are present in eye lens: Evidence for all
major erythrocyte membrane components in same non-erythroid cell. Biochem Biophys Res Commun 149:266, 1987 162. Kuwabara T: Microtubules in the lens. Arch Ophthalmol 79:189, 1968 163. Piatigorsky J: Lens cell elongation in vitro and microtubules. Ann NY Acad Sci 253:333, 1975 164. Rafferty NS, Scholz DL, Goldberg M, Lewyckyj M: Immunocytochemical evidence for an actin-myosin system in lens epithelial
cells. Exp Eye Res 51:591, 1990 165. Ireland M, Lieska N, Maisel H: Lens actin: Purification, localization. Exp Eye Res 37:393, 1983 166. Kibbelaar MA, Ramaekers FC, Ringens PJ et al: Is actin in eye lens a possible factor in visual accommodation? Nature 285:506, 1980 167. Rafferty NS, Scholz DL: Comparative study of actin filament patterns in lens epithelial cells. Are
these determined by the mechanisms of lens accommodation? Curr Eye Res 8:569, 1989 168. Alcala J, Maisel H: Biochemistry of lens plasma membranes and cytoskeleton. In
Maisel H (ed): The Ocular Lens: Structure, Function, and Pathology. New
York: Marcel Dekker, 1985 169. Kam Z, Volberg T, Geiger B: Mapping of adherens junction components using microscopic resonance energy
transfer imaging. J Cell Sci 108:1051, 1995 170. Lo WK, Mills A, Kuck JF: Actin filament bundles are associated with fiber gap junctions in the primate
lens. Exp Eye Res 58:189, 1994 171. Sussman MA, McAvoy JW, Rudisill M et al: Lens tropomodulin: Developmental expression during differentiation. Exp Eye Res 63:223, 1996 172. Lo WK, Shaw AP, Wen XJ: Actin filament bundles in cortical fiber cells of the rat lens. Exp Eye Res 65:691, 1997 173. Ramaekers FC, Osborn M, Schimid E et al: Identification of the cytoskeletal proteins in lens-forming cells, a special
epithelioid cell type. Exp Cell Res 127:309, 1980 174. Sredy J, Roy D, Spector A: Identification of two of the major phosphorylated polypeptides of the bovine
lens utilizing a lens cAMP-dependent protein kinase system. Curr Eye Res 3:1423, 1984 175. Sandilands A, Prescott AR, Carter JM et al: Vimentin and CP49/filensin form distinct networks in the lens which are
independently modulated during lens fibre cell differentiation. J Cell Sci 108:1397, 1995 176. Sax CM, Farrell FX, Zehner ZE, Piatigorsky J: Regulation of vimentin gene expression in the ocular lens. Dev Biol 139:56, 1990 177. Krimpenfort PJ, Schaart G, Peiper FR et al: Tissue-specific expression of a vimentin-desmin hybrid gene in transgenic
mice. EMBO J 7:941, 1988 178. Hatfield JS, Skoff RP, Maisel H et al: The lens epithelium contains glial fibrillary acidic protein (GFAP). J Neuroimmunol 8:347, 1985 179. Boyer S, Maunoury R, Gomes D et al: Expression of glial fibrillary acidic protein and vimentin in mouse lens
epithelial cells during development in vivo and during proliferation
and differentiation in vitro: Comparison with the developmental appearance
of GFAP in the mouse central nervous system. J Neurosci Res 27:55, 1990 180. FitzGerald PG, Gottlieb W: The Mr 115-kd fiber cellspecific protein is a component of the lens cytoskeleton. Curr Eye Res 8:801, 1989 181. FitzGerald PG, Casselman J: Discrimination between the lens fiber cell 114-kd cytoskeletal protein
and alpha-actinin. Curr Eye Res 9:873, 1990 182. Goulielmos G, Gounari F, Remington S et al: Filensin and phakinin form a novel type of beaded intermediate filaments
and coassemble de novo in cultured cells. J Cell Biol 132:643, 1996 183. Ireland M, Maisel H: Phosphorylation of chick lens proteins. Curr Eye Res 3:961, 1984 184. Hess JF, Casselman JT, FitzGerald PG: Gene structure and cDNA sequence identify the beaded filament protein CP49 as
a highly divergent type I intermediate filament protein. J Biol Chem 271:6729, 1996 185. Mathias RT, Rae JL: Transport properties of the lens. Am J Physiol 249:C181, 1985 186. Georgatos SD, Gounari F, Goulielmos G, Aebi U: To bead or not to bead? Lens-specific intermediate filaments revisited. J Cell Sci 110:2629, 1997 187. Carter JM, Hutcheson AM, Quinlan RA: In vitro studies on the assembly properties of the lens proteins CP49 and CP115: Coassembly
with alpha-crystallin but not with vimentin. Exp Eye Res 60:181, 1995 188. Nicholl ID, Quinlan RA: Chaperone activity of alphacrystallins modulates intermediate filament
assembly. EMBO J 13:945, 1994 189. Huang CH, Chen Y, Reid M, Ghosh S: Genetic recombination at the human RH locus: A family study of the red-cell
Evans phenotype reveals a transfer of exons 2–6 from the RHD
to the RHCE gene. Am J Hum Genet 59:825, 1996 190. Eiberg H, Nielsen LS, Klausen J et al: Linkage between serum cholinesterase 2 (CHE2) and gamma-crystallin gene
cluster (CRYG): Assignment to chromosome 2. Clin Genet 35:313, 1989 191. Marner E, Rosenberg T, Eiberg H: Autosomal dominant congenital cataract: Morphology and genetic mapping. Acta Ophthalmol 67:151, 1989 192. Armitage MM, Kivlin JD, Ferrell RE: A progressive early-onset cataract gene maps to human chromosome 17q24. Nat Genet 9:37, 1995 193. Seri M, Cusano R, Forabosco P et al: Genetic mapping to 10q23 3-q24 2, in a large Italian pedigree, of a new
syndrome showing bilateral cataracts, gastroesophageal reflux and spastic
paraparesis with amyotrophy. Am J Hum Genet 64:586, 1999 194. Ogata H, Okubo Y, Akabane T: Phenotype i associated with congenital cataract in Japanese. Transfusion 19:166, 1979 195. Macdonald EB, Douglas R, Harden PA: A caucasian family with the i phenotype and congenital cataracts. Vox Sang 44:322, 1983 196. Yamaguchi H, Okubo Y, Tanaka M: A note on possible close linkage between the Ii blood locus and a congenital
cataract locus. Proc Jpn Acad 48:625, 1972 197. Levi S, Girelli D, Perrone F et al: Analysis of ferritins in lymphoblastoid cell lines in the lens of subjects
with hereditary hyperferritinemia-cataract syndrome. Blood 91:4180, 1998 198. Elsas FJ, Maumenee IH, Kenyon KR, Yoder F: Familial aniridia with preserved ocular function. Am J Ophthalmol 83:718, 1977 199. Traboulsi EI, Weinberg RJ: Familial congenital cornea guttata with anterior polar cataracts. Am J Ophthalmol 108:123, 1989 200. Moller HU: Granular corneal dystrophy Groenouw type I: Clinical aspects and treatment. Acta Ophthalmol 68:384, 1990 201. Gitter KA, Rothschild H, Waltman DD et al: Dominantly inherited peripheral retinal neovascularization. Arch Ophthalmol 96:1601, 1978 202. O'Donnell FE, Pappas HR: Autosomal dominant foveal hypoplasia and presenile cataracts: A new syndrome. Arch Ophthalmol 100:279, 1982 203. Alexander RL, Shea M: Wagner's disease. Arch Ophthalmol 74:310, 1965 204. Beaumont C, Leneuve P, Devaux I et al: Mutation in the iron responsive element of the L-ferritin mRNA in a family
with dominant hyperferritinaemia and cataract. Nat Genet 11:444, 1995 205. Kafer O: Dominant vererbte Spaltung des Pigmentblattes von Iris und Ciliarkoepfer
mit consekutiver Microphakie, Ectopia lentis und Cataract. Graefes Arch Klin Exp Ophthal 202:133, 1977 206. Hittner HM, Kretzer FL, Antoszyk JH et al: Variable expressivity of autosomal dominant anterior segment mesenchymal
dysgenesis in six generations. Am J Ophthalmol 93:57, 1982 207. Friedmann MW, Wright ES: Hereditary microcornea and cataract in 5 generations. Am J Ophthalmol 35:1017, 1952 208. Capella JA, Kaufman HE, Lill FJ, Cooper G: Hereditary cataracts and microphthalmia. Am J Ophthalmol 56:454, 1963 209. Cassady JR, Light A: Familial persistent pupillary membranes. Arch Ophthalmol 58:438, 1957 210. Heckenlively J: The frequency of posterior subcapsular cataract in the hereditary retinal
degenerations. Am J Ophthalmol 93:733, 1982 211. Berson EL, Rosner B, Simonoff E: Risk factors for genetic typing, detection in retinitis pigmentosa. Am J Ophthalmol 89:763, 1980 212. Hirose T, Lee KY, Schepens CL: Snowflake degeneration in hereditary vitreoretinal degeneration. Am J Ophthalmol 77:143, 1974 213. Kaufman SJ, Goldberg MF, Orth DH et al: Autosomal dominant vitreoretinochoroidopathy. Arch Ophthalmol 100:272, 1982 214. Stock EL, Kielar RA: Primary familial amyloidosis of the cornea. Am J Ophthalmol 82:266, 1976 215. Favre M: A propos de deux cas de degenerescence hyaloideoretinienne. [Two cases
of hyaloid-retinal degeneration.] Ophthalmologica 135:604, 1958 216. Alstrom CH: Heredo-retinopathia congenitalis monohybrida recessiva autosomalis: A genetic
statistical study in clinical collaboration with Olof Olson. Hereditas 43:1, 1957 217. Temtamy SA, Shalash BA: Genetic heterogeneity of the syndrome: Microphthalmos
with congenital cataract. Birth Defects Orig Art Ser. X(4):292, 1974 218. Warburg M: Norrie's disease: A new hereditary bilateral pseudotumour of the retina. Arch Ophthalmol 39:757, 1961 219. Mendelian Inheritance in Man. Baltimore: The Johns Hopkins University Press, 1992 220. Heck AF: Presumptive X-linked intermediate transmission of retinal degenerations: Variations, coincidental
occurrence with ataxia in a large family. Arch Ophthalmol 70: 143, 1963 221. Wellesley D, Carman P, French N, Goldblatt J: Cataracts, aberrant oral frenula, growth retardation: A new autosomal dominant
syndrome. Am J Med Genet 40:341, 1991 222. Stromgen E, Dalby A, Dalby MA, Ranheim B: Cataracts, deafness, cerebellar ataxia, psychosis, and dementia—a
new syndrome. Acta Neurol Scand 43(suppl):261, 1970 223. Spranger JW, Opitz JM, Bidder U: Heterogeneity of chondrodysplasia punctata. Humangenetik 11:190, 1971 224. Nadol JB Jr, Burgess B: Cochleosaccular degeneration of the inner ear and progressive cataracts
inherited as an autosomal dominant trait. Laryngoscope 92:1028, 1982 225. Russo G, Mollica F, Mazzone D, Santonocito B: Congenital lactose intolerance of gastrogenic origin associated with cataracts. Acta Paediat Scand 63:457, 1974 226. Moore WT, Federman DD: Familial dwarfism and stiff joints. Arch Intern Med 115:398, 1965 227. Cochat P, Guibaud P, Garcia-Torres R et al: Diffuse leiomyomatosis in Alport syndrome. Pediatrics 113:339, 1988 228. Flynn P, Aird RB: A neuroectodermal syndrome of dominant inheritance. J Neurol Sci 2:161, 1965 229. Francois J: A new syndrome: Dyscephalia with bird face, dental anomalies, nanism, hypotrichosis, cutaneous
atrophy, microphthalmia, and congenital cataract. Arch Ophthalmol 60:842, 1958 230. Witkopf CJ, White JG, King RA et al: Hereditary mucoepithelial dysplasia: A disease apparently of desmosome
and gap junction formation. Am J Hum Genet 31:414, 1979 231. Zayid I, Farraj S: Familial histiocytic dermatoarthritis: A new syndrome. Am J Med 54:793, 1973 232. Carney RG: Incontinentia pigmenti: A world statistical analysis. Arch Dermatol 112:535, 1976 233. Tyni T, Kivela T, Lappi M et al: Ophthalmologic findings in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency
caused by the G1528C mutation: A new type of hereditary metabolic
chorioretinopathy. Ophthalmology 105:810, 1998 234. Kniest W, Leiber B: Kniest's syndrome. Monatsschr Kinderheilkd 125:970, 1977 235. Tessler HH, Apple DJ, Goldberg MF: Ocular findings in a kindred with Kyrle disease. Arch Ophthalmol 90:278, 1973 236. Pepin B, Mikol J, Goldstein B et al: Familial mitochondrial myopathy with cataract. J Neurol Sci 45:191, 1980 237. Walker BA: Osteopathia striata with cataracts and deafness. Birth Defects Orig Art Ser. 4:295, 1969 238. Marshall D: Ectodermal dysplasia. Report of a kindred with ocular abnormalities and
hearing defect. Am J Ophthalmol 45:143, 1958 239. Beighton P, Goldberg L, Op't Hof J: Dominant inheritance of multiple epiphyseal dysplasia, myopia, and deafness. Clin Genet 14:173, 1978 240. Ashizawa T, Hejtmancik JF, Liu J et al: Diagnostic value of ophthalmologic findings in myotonic dystrophy: Comparison
with risks calculated by haplotype analysis of closely linked restriction
fragment length polymorphisms. Am J Med Genet 42:55, 1992 241. Quintanilla E, Rodrigo A, Temino MA et al: [Nail-patella syndrome with ocular involvement. Study of 5 generations.] Actas Dermosifiliogr 72:415, 1981 242. Kaiser-Kupfer MI, Freidlin V, Datiles MB et al: The association of posterior capsular lens opacities with bilateral acoustic
neuromas in patients with neurofibromatosis type 2. Arch Ophthalmol 107:541, 1989 243. Garcin R, Delthil S, Man HX, Chimenes H: Sur une affection heredo-familiale associant cataracte, atrophie optique, signes
extra-pyramidaux et certains stigmates de la maladie de Friedreich. (Sa
position nosologique par rapport au syndrome de Behr, au syndrome
de Marinesco-Sjogren et a la maladie de Friedreich avec signes
oculaires.) Rev Neurol 104:373, 1961 244. DeBusk FL: The Hutchinson-Gilford progeria syndrome. J Pediatr 80:697, 1972 245. Shprintzen RJ, Wang F, Goldberg R, Marion R: The expanded velo-cardio-facial syndrome (VCF): Additional features of
the most common clefting syndrome. Am J Hum Genet 37:A77, 1985 246. Vaca G, Ibarra B, Bracamontes M et al: Red blood cell sorbitol dehydrogenase deficiency in a family with cataracts. Hum Genet 61:338, 1982 247. Neugebauer H: Spalthand und fus mit familiarer besonderheit. Z Orthop 95:500, 1962 248. Seery CM, Pruett RC, Liberfarb RM, Cohen BZ: Distinctive cataract in the Stickler syndrome. Am J Ophthalmol 110:143, 1990 249. Goldstein JH, Hutt AE: Trichomegaly, cataract, and hereditary spherocytosis in two siblings. Am J Ophthalmol 73: 333, 1972 250. McKusick VA, Weilbaecher RG, Gragg GW: Recessive inheritance of a congenital malformation syndrome. JAMA 204:113, 1968 251. Dionisi Vici C, Sabetta G, Gambarara M et al: Agenesis of the corpus callosum, combined immunodeficiency, bilateral cataract, and
hypopigmentation in two brothers. Am J Med Genet 29:1, 1988 252. Lyon G, Arita F, Le Galloudec E et al: A disorder of axonal development, necrotizing myopathy, cardiomyopathy, and
cataracts: A new familial disease. Ann Neurol 27:193, 1990 253. Riise R: Visual function in Laurence-Moon-Bardet-Biedl syndrome. Acta Ophthalmol Suppl (Copenh) 182:128, 1987 254. Sengers RCA, ter Haar BGA, Trijbels JMF et al: Congenital cataract and mitochondrial myopathy of skeletal and heart muscle
associated with lactic acidosis after exercise. J Pediatr 86:873, 1975 255. Seland JH, Slagsvold JE: The ultrastructure of lens and iris in cerebrotendinous xanthomatosis. Acta Ophthalmol 55:201, 1977 256. Grizzard WS, O'Donnell JJ, Carey JC: The cerebro-oculo-facio-skeletal syndrome. Am J Ophthalmol 89:293, 1980 257. Pearce WG: Ocular and genetic features of Cockayne's syndrome. Can J Ophthal 7:435, 1972 258. Pinkerton OD: Cataract associated with congenital ichthyosis. Arch Ophthalmol 60:393, 1958 259. Crome L, Duckett S, Franklin AW: Congenital cataracts, renal tubular necrosis, and encephalopathy in two
sisters. Arch Dis Child 38:505, 1963 260. Sanner G: The dysequilibrium syndrome. A genetic study. Neuropadiatrie 4:403, 1973 261. Gitzelmann R: Hereditary galactokinase deficiency, a newly recognized cause of juvenile
cataracts. Pediatr Res 1:14, 1967 262. Loos H, Roos D, Weening R, Houwerzijl J: Familial deficiency of glutathione reductase in human blood cells. Blood 48:53, 1976 263. Caspersen I, Warburg M: Hallermann-Streiff syndrome. Acta Ophthalmol 46:358, 1968 264. Pagon RA, Clarren SK, Milam DF Jr, Hendrickson AE: Autosomal recessive eye and brain anomalies: Warburg syndrome. J Pediatr 102:542, 1983 265. Spaeth GL, Barber GW: Homocystinuria—its ocular manifestations. J Pediatr Ophthalmol 3:42, 1966 266. Gold GN, Hogenhuis LAH: Hypertrophic interstitial neuropathy and cataracts. Neurology 18:526, 1968 267. Lubinsky MS: Cataracts and testicular failure in three brothers. Am J Med Genet 16:149, 1983 268. Buyse M, Bull MJ: A syndrome of osteogenesis imperfecta, microcephaly, and
cataracts. Birth Defects Orig Art Ser. XIV(6B):95, 1978 269. Arbisser AI, Murhree AL, Garcia CA, Howell RR: Ocular findings in mannosidosis. Am J Ophthalmol 82:465, 1976 270. Chess J, Albert DM: Ocular pathology of the Majewski syndrome. Br J Ophthal 66:736, 1982 271. Herva R, von Wendt L, von Wendt G et al: A syndrome with juvenile cataract, cerebellar atrophy, mental retardation, and
myopathy. Neuropediatrics 18:164, 1987 272. Martsolf JT, Hunter AGW, Haworth JC: Severe mental retardation, cataracts, short stature, and primary hypogonadism
in two brothers. Am J Med Genet 1:291, 1978 273. Hoffmann G, Gibson KM, Brandt IK et al: Mevalonic aciduria—an inborn error of cholesterol and nonsterol isoprene
biosynthesis. N Engl J Med 314:1610, 1986 274. Lundberg PO: Hereditary myopathy, oligophrenia, cataract, skeletal abnormalities, and
hypergonadotropic hypogonadism: A new syndrome. Acta Genet Med Gemellol 23:245, 1974 275. Cremers CWRJ, ter Haar BGA, Van Rens TJG: The Nathalie syndrome. A new hereditary syndrome. Clin Genet 8:330, 1975 276. Neu RL, Kajii T, Gardner LI, Nagyfy SF: A lethal syndrome of microcephaly with multiple congenital anomalies in
three siblings. Pediatrics 47:610, 1971 277. Thomas PK, Abrams JD, Swallow D, Stewart G: Sialidosis type I: Cherry red spot/myoclonus syndrome with sialidase deficiency
and altered electrophoretic mobility of some enzymes known to
be glycoproteins. I. Clinical findings. J Neurol Neurosurg Psychiat 42:873, 1979 278. Williams ML, Koch TK, O'Donnell JJ et al: Ichthyosis and neutral lipid storage disease. Am J Med Genet 20:711, 1985 279. Salih MAM, Bender DA, McCreanor GM: Lethal familial pellagra-like skin lesion associated with neurologic and
developmental impairment and the development of cataracts. Pediatrics 76:787, 1985 280. Kwashima H, Kawano M, Masaki A, Sato T: Three cases of untreated classical PKU: A report on cataracts and brain
calcification. Am J Med Genet 29:89, 1988 281. Fairly KF, Leighton PW, Kincaid-Smith P: Familial visual defects associated with polycystic kidney and medullary
sponge kidney. Br Med J 1:1060, 1963 282. Preus M, Fraser FC, Wigglesworth JW: An oculocerebral hypopigmentation syndrome. J Genet Hum 31:323, 1983 283. Folz SJ, Trobe JD: The peroxisome and the eye. Surv Ophthalmol 35:353, 1991 284. Jones KL: Smith's Recognizable Patterns of Human Malformation. Philadelphia: WB
Saunders, 1988 285. Rothmund A: Uber Cataracte in Verbindung mit einer eigenthuemlichen Hautdegeneration. Graefes Arch Klin Exp Ophthal 14:159, 1868 286. Mollica F, Pavone L, Antener I: Short stature, mental retardation, and ocular alterations in three siblings. Helv Paediat Acta 27:463, 1972 287. Cotlier E, Rice P: Cataracts in the Smith-Lemli-Opitz syndrome. Am J Ophthalmol 72:955, 1971 288. Adams CW, Nance WE: Persistent tachycardia, paroxysmal hypertension, and seizures: Association
with hyperglycinuria, dominantly inherited microphthalmia, and cataracts. JAMA 202:525, 1967 289. Toriello HV, Horton WA, Oostendorp A et al: An apparently new syndrome of microcephalic primordial dwarfism, cataracts. Am J Med Genet 25:1, 1986 290. Hallgren B: Retinitis pigmentosa combined with congenital deafness: With vestibulo-cerebellar
ataxia and mental abnormality in a proportion of cases. Acta Psychiat Neurol Scand 34 (suppl. 138):9, 1959 291. Epstein CJ, Martin GM, Schultz AL, Motulsky AG: Werner's syndrome: A review of its symptomatology, natural history, pathologic
features, genetics, and relationship to the natural aging
process. Medicine 45:177, 1966 292. Tso MO, Fine BS, Thorpe HE: Kayser-Fleischer ring and associated cataract in Wilson's disease. Am J Ophthalmol 79:479, 1975 293. Fitch N: Albright's hereditary osteodystrophy: A review. Am J Med Genet 11:11, 1982 294. Arnott EJ, Crawford MD, Toghill PJ: Anterior lenticonus and Alport's syndrome. Br J Ophthal 30:390, 1966 295. Scher NA, Letson RD, Desnick RJ: The ocular manifestations in Fabry's disease. Arch Ophthalmol 97:671, 1979 296. Orzalesi N, Sorcinelli R, Guiso G: Increased incidence of cataracts in male subjects deficient in glucose-6-phosphate
dehydrogenase. Arch Ophthalmol 99:69, 1981 297. Carney RG: Incontinentia pigmenti, a world statistical analysis. Arch Dermatol 112:535, 1976 298. Herrmann H, Opitz JM: The Lenz microphthalmia syndrome. Birth Defects Orig
Art Ser. V(2):138, 1969 299. Wadelius C, Fagerholm P, Pettersson U, Anneren G: Lowe oculocerebrorenal syndrome: DNA-based linkage of the gene to X124-q26, using
tightly linked flanking markers and the correlation to lens
examination in carrier diagnosis. Am J Hum Genet 44:241, 1989 300. Nance WE, Warburg M, Bixler D, Helveston EM: Congenital X-linked cataract, dental
anomalies, and brachymetacarpalia. Birth Defects Orig Art Ser. X(4):285, 1974 301. Mirhosseini SA, Holmes LB, Walton DS: Syndrome of pigmentary retinal degeneration, cataract, microcephaly, and
severe mental retardation. J Med Gen 9:193, 1972 302. Winsnes A, Monn E, Stokke O, Feyling T: Congenital persistent proximal type renal tubular acidosis in two brothers. Acta Paediat Scand 68:861, 1979 303. Happle R: Cataracts as a marker of genetic heterogeneity in chondrodysplasia punctata. Clin Genet 19:64, 1981 304. Reese PD, Tuck-Miller CM, Maumenee IH: Autosomal dominant congenital cataract associated with chromosomal translocation [t(3;4)(p26 2;p15)]. Arch Ophthalmol 105: 1382, 1987 305. Moross T, Vaithilingam SS, Styles S, Gardner HA: Autosomal dominant anterior polar cataracts associated with a familial 2;14 translocation. J Med Genet 21:52, 1984 306. Yokoyama Y, Narahara K, Tsuji K et al: Autosomal dominant congenital cataract and microphthalmia associated with
a familial t(2;16) translocation. Hum Genet 90:177, 1992 307. Monaghan KG, Van DD, Wiktor A, Feldman GL: Cytogenetic and clinical findings in a patient with a deletion of 16q23 1: First
report of bilateral cataracts and a 16q deletion. Am J Med Genet 73:180, 1997 308. Rubin SE, Nelson LB, Pletcher BA: Anterior polar cataract in two sisters with an unbalanced 3;18 chromosomal
translocation. Am J Ophthalmol 117:512, 1994 309. Petit P, Devriendt K, Vermeesch JR et al: Unusual de novo t(13;15)(q12 1;p13) translocation leading to complex mosaicism
including jumping translocation. Ann Genet 41:22, 1998 310. Schlote T, Volker M, Knorr M, Thiel HJ: [Lens coloboma, lens dislocation in Stickler (Marshall) syndrome.] Klin Monatsbl Augenheilkd 210:227, 1997 311. Mehrotra AS, Solanki N, Sabharwal KK: Bilateral coloboma of lens in Marfan's syndrome. Indian J Ophthalmol 33:201, 1985 312. Jensen AD, Cross HE, Paton D: Ocular complications in the Weill-Marchesani syndrome. Am J Ophthal 77:261, 1974 313. Colville DJ, Savige J: Alport syndrome. A review of the ocular manifestations. Ophthalmic Genet 18:161, 1997 314. Mir S, Wheatley HM, Hussels IE et al: A comparative histologic study of the fibrillin microfibrillar system in
the lens capsule of normal subjects and subjects with Marfan syndrome. Invest Ophthalmol Vis Sci 39:84, 1998 315. Mudd SH, Skovby F, Levy HL et al: The natural history of homocystinuria due to cystathionine beta-synthase
deficiency. Am J Hum Genet 37:1, 1985 316. Wirtz MK, Samples JR, Kramer PL et al: Weill-Marchesani syndrome—possible linkage of the autosomal dominant
form to 15q21.1. Am J Med Genet 65:68, 1996 317. Goh A, Lim KW: Sulphite oxidase deficiency—a report of two siblings. Singapore Med J 38:391, 1997 318. Tsipouras P, Del Mastro R, Sarfarazi M et al: Genetic linkage of the Marfan syndrome, ectopia lentis, and congenital
contractural arachnodactyly to the fibrillin genes on chromosomes 15 and 5. N Engl J Med 326:906, 1992 319. Dobson V, Teller DY: Visual acuity in human infants: A review and comparison of behavioral and
electrophysiological studies. Vision Res 18:1469, 1978 320. Atkinson J, Braddick O: Assessment of visual acuity in infancy and early childhood. Acta Ophthalmol Suppl (Copenh) 157:18, 1983 321. Weisel TN, Hubel DH: Comparison of the effects of unilateral and bilateral eyelid closure on
cortical responses in kittens. J Neurophysiol 28:1029, 1965 322. Harwerth RS, Smith EL III, Paul AD et al: Functional effects of bilateral form deprivation in monkeys. Invest Ophthalmol Vis Sci 32:2311, 1991 323. Robb RM, Mayer DL, Moore BD: Results of early treatment of unilateral congenital cataracts. J Pediatr Ophthalmol Strabismus 24:178, 1987 324. Nelson LB: Diagnosis and management of cataracts in infancy and childhood. Ophthalmic Surg 15:688, 1984 325. Gelbart SS, Hoyt CS, Jastrebski G, Marg E: Long-term visual results in bilateral congenital cataracts. Am J Ophthal 93:615, 1982 326. Burns EC, Jones RB: Long-term management of congenital cataracts. Arch Dis Child 60:322, 1985 |