1. Delaye M, Tardieu A: Short range order of crystallin proteins accounts for eye lens transparency. Nature 302:415, 1983 2. Phelps Brown N, Bron AJ: Lens Disorders—A Clinical Manual of Cataract
Diagnosis. Oxford: Butterworth-Heinemann, 1996 3. Mathias RT, Rae JL, Baldo GJ: Physiological properties of the normal lens. Physiol Rev 77:21, 1997 4. Duncan G, Bushell AR: Relationships between colour, sodium and protein content in individual
senile cataractous lenses. Ophthal Res 11:397, 1979 5. Jedziniak JA, Nicoli DF, Baram H et al: Quantitative verification of the existence of high-molecular-weight protein
aggregates in the intact normal human lens by light-scattering spectroscopy. Invest Ophthalmol Visual Sci 17:51, 1978 6. Al-Ghoul KJ, Novak LA, Kuszak JR: The structure of posterior subcapsular cataracts in the Royal College of
Surgeons (RCS) rats. Exp Eye Res 67:163, 1998 7. Spector A: Oxidative stress-induced cataract: Mechanisms of action. FASEB J 9:1173, 1995 8. Giblin FJ, Padgaonkar VA, Leverenz VR et al: Nuclear light scattering, disulfide formation and membrane damage in lenses
of older guinea pigs treated with hyperbaric oxygen. Exp Eye Res 60:219, 1995 9. Hiller R, Sperduto RD, Ederer F: Epidemiologic associations with nuclear, cortical and posterior subcapsular
cataracts. Am J Epidemiol 124:916, 1986 10. Taylor HR, West SK, Rosenthal FS: Effect of ultraviolet radiation on cataract formation. N Engl J Med 319:1429, 1988 11. Spector A: The lens and oxidative stress. In Sies H (ed): Oxidative Stress: Oxidants
and Antioxidants, p 529. London, Academic Press, 1991 12. Cheng H-M, Chylack LT Jr: Lens metabolism. In Maisel H (ed): The Ocular
Lens, p 223. New York: Marcel-Dekker, 1985 13. Dillon J, Zheng L, Merriam JC: The optical properties of the anterior segment of the eye: Implications
for cortical cataract. Exp Eye Res 68:785, 1999 14. Zigler JS Jr, Goosey JD: Photosensitized oxidation in the ocular lens: Evidence for photosensitizers
endogenous to the human lens. Photochem Photobiol 33:869, 1981 15. Linetsky M, Ortwerth BJ: Quantitation of the singlet oxygen produced by UV-A irradiation of human
lens proteins. Photochem Photobiol 65:522, 1997 16. Spector A, Garner WH: Hydrogen peroxide and human cataract. Exp Eye Res 33:673, 1981 17. Bleau G, Giasson C, Brunette I: Measurement of hydrogen peroxide in biological samples containing high
levels of ascorbic acid. Anal Biochem 263:13, 1998 18. Zigler JS Jr, Jernigan HM Jr, Garland DL et al: The effects of “oxygen radicals” generated in the medium on
lenses in organ culture: Inhibition of damage by chelated iron. Arch Biochem Biophys 241:163, 1985 19. Hanson SRA, Chen AA, Smith JB et al: Thiolation of the γB-crystallin in intact bovine lens exposed to
hydrogen peroxide. J Biol Chem 274:4735, 1999 20. Reddy VN: Metabolism of glutathione in the lens. Exp Eye Res 11:310, 1971 21. Giblin FJ, McCready JP, Kodama T et al: A direct correlation between the levels of ascorbic acid and H2O2 in aqueous humor. Exp Eye Res 38:87, 1984 22. Giblin FJ, Nies DE, Reddy VN: Stimulation of the hexose monophosphate shunt in rabbit lens in response
to the oxidation of glutathione. Exp Eye Res 33:289, 1981 23. Yang Y, Spector A, Ma W et al: The effect of catalase amplification on immortal lens epithelial cell lines. Exp Eye Res 67:647, 1998 24. Reddy VN, Lin LK, Ho YS et al: Peroxide-induced damage in lenses of transgenic mice with deficient and
elevated levels of glutathione peroxidase. Ophthalmologica 211:192, 1997 25. Wang RR, Kuszak JR, Ma W: The effect of aging on the lenses of glutathione peroxidase-1 knockout. Invest Ophthalmol Vis Sci 40:5877, 1999 26. Lou MF: Protein-thiol mixed disulfides and thioltransferase in the lens. In
Green K, Edelhauser HF, Hackett RB, et al (eds): Advances in Ocular
Toxicology, p 27. New York: Plenum Press, 1997 27. Horwitz J: Alpha-crystallin can function as a molecular chaperone. Proc Natl Acad Sci USA 89:10449, 1992 28. Benedek GB: Cataract as a protein condensation disease. Invest Ophthalmol Vis Sci 38:1911, 1997 29. Tanaka T, Benedek GB: Observation of protein diffusivity in intact human and bovine lenses with
application to cataract. Invest Ophthalmol 14:449, 1975 30. Pande J, Ogun O, Nath C et al: Suppression of phase separation in bovine γIV-crystallin solutions: Effect
of modification by charged versus uncharged polar groups. Exp Eye Res 57: 257, 1993 31. Clark JI, Carper D: Phase separation in lens cytoplasm is genetically linked to cataract formation
in the Philly mouse. Proc Natl Acad Sci USA 84:122, 1987 32. Clark JI, Livesey JC, Steele JE: Delay or inhibition of rat lens opacification using pantethine and WR-77913. Exp Eye Res 62:75, 1996 33. Robison WG Jr, Houlder N, Kinoshita JH: The role of lens epithelium in sugar cataract formation. Exp Eye Res 50: 641, 1990 34. Horwitz J, Dovrat A, Straatsma BR et al: Glutathione reductase in the human lens epithelium. Curr Eye Res 6:1249, 1987 35. Kantorow M, Kays T, Horwitz J et al: Differential display detects altered gene expression between cataractous
and normal human lenses. Invest Ophthalmol Vis Sci 39:2344, 1998 36. Reddy VN, Lin L-R, Arita T et al: Crystallins and their synthesis in human lens epithelial cells in tissue
culture. Exp Eye Res 47:465, 1988 37. Andley UP, Rhim JS, Chylack LT Jr et al: Propagation and immortalization of human lens epithelial cells in culture. Invest Ophthalmol Vis Sci 35:2094, 1994 38. Potts JD, Dagle MM, Walder JA et al: Epithelial-mesenchymal transformation of embryonic cardiac endothelial
cells is inhibited by a modified antisense oligonucleotide to transforming
growth factor β3. Proc Natl Acad Sci USA 88: 1516, 1991 39. Hales AM, Chamberlain CG, McAvoy JW: Cataract induction in lenses cultured with transforming growth factor β. Invest Ophthalmol Vis Sci 36:1709, 1995 40. Srinivasan Y, Lovicu FA, Overbeck PA: Lens-specific expression of transforming growth factor β1 in transgenic
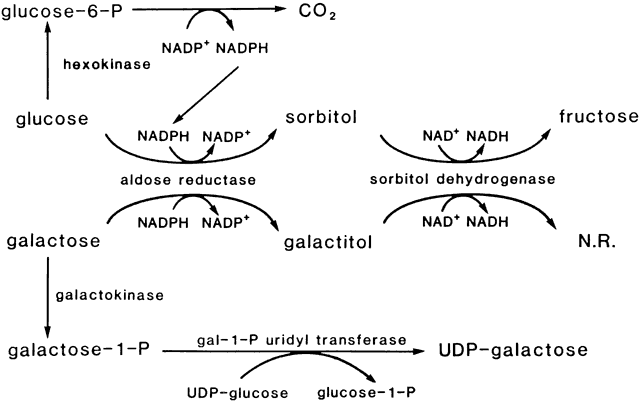
mice causes anterior subcapsular cataracts. J Clin Invest 101: 625, 1998 41. Hales AM, Chamberlain CG, Dreher B et al: Introvitreal injection of TGFβ induces cataract in rats. Invest Ophthalmol Vis Sci 40:3231, 1999 42. Hales AM, Chamberlain CG, Murphy CR et al: Estrogen protects lenses against cataract induced by TGFβ. J Exp Med 185:273, 1997 43. van Heyningen R: Formation of polyols by the lens of the rat with sugar cataract. Nature 184:194, 1959 44. Kinoshita JH, Datiles MB, Kador PF et al: Aldose reductase and diabetic
eye complications. In Rifkin H, Porte D (eds): Diabetes Mellitus: Theory
and Practice, 4th ed, p 264. New York: Elsevier Science, 1990 45. Kinoshita JH: Mechanism of cataract formation. Invest Ophthalmol 13:713, 1974 46. Datiles M, Fukui H, Kuwabara T et al: Galactose cataract prevention with Sorbinil, an aldose reductase inhibitor: A
light microscopic study. Invest Ophthalmol Vis Sci 22:174, 1982 47. Kinoshita JH, Kador P, Datiles M: Aldose reductase in diabetic cataracts. JAMA 246:259, 1981 48. Varma SD, Kinoshita JH: The absence of cataracts in mice with congenital hyperglycemia. Exp Eye Res 19:577, 1974 49. Lee AY, Chung SK, Chung SS: Demonstration that polyol accumulation is responsible for diabetic cataract
by the use of transgenic mice expressing the aldose reductase gene
in the lens. Proc Natl Acad Sci USA 92:2780, 1995 50. Datiles M, Fukui H: Cataract prevention in diabetic Octodon degus using Pfizer's sorbinil. Curr Eye Res 8:233, 1989 51. Kinoshita JH: Aldose reductase and the diabetic eye. Am J Ophthalmol 102:685, 1986 52. Harding JJ: Can cataract be prevented? Eye 13:454, 1999 53. Shearer TR, David LL, Anderson RS et al: Review of selenite cataract. Curr Eye Res 11:357, 1992 54. Mitton KP, Hess JL, Bunce GE: Causes of decreased phase transition temperature in selenite cataract model. Invest Ophthalmol Vis Sci 36:914, 1995 55. Calvin HI, Medvedovsky C, Worgul BV: Near-total glutathione depletion and age-specific cataracts induced by
buthionine sulfoximine in mice. Science 233:553, 1986 56. Calvin KI, Zhu G-P, Wu J-X et al: Progression of mouse buthionine sulfoximine cataracts in vitro is inhibited by thiols or ascorbate. Exp Eye Res 65:341, 1997 57. Giblin FJ, Padgaonkar VA, Leverenz VR et al: Nuclear light scattering, disulfide formation and membrane damage in lenses
of older guinea pigs treated with hyperbaric oxygen. Exp Eye Res 60:219, 1995 58. Padgaonkar VA, Lin L-R, Leverenz VR et al: Hyperbaric oxygen in vivo accelerates the loss of cytoskeletal proteins and MIP26 in guinea pig
lens nucleus. Exp Eye Res 68:493, 1999 59. Iwata S, Kinoshita JH: Mechanism of development of hereditary cataract in mice. Invest Ophthalmol 10:504, 1960 60. Fukui HN, Merola LO, Kinoshita JH: A possible cataractogenic factor in the Nakano mouse lens. Exp Eye Res 26: 477, 1978 61. Kador PF, Fukui HN, Fukushi S et al: Philly mouse: A new model of hereditary cataract. Exp Eye Res 30:59, 1980 62. Chambers C, Russell P: Deletion mutation in an eye lens β-crystallin. J Biol Chem 266:6742, 1991 63. Kramer P, Yount J, Michell T et al: A second gene for cerulean cataracts maps to the beta-crystallin region
on chromosome 22. Genomics 35:539, 1996 64. Rodriguez IR, Gonzalez P, Zigler JS Jr et al: A guinea pig hereditary cataract contains a splice site deletion in a crystallin
gene. Biochem Biophys Acta 1180:44, 1992 65. Rao PV, Zigler JS Jr: Mutant zeta-crystallin from guinea pig hereditary cataracts has altered
structural and enzymatic properties. Exp Eye Res 54:627, 1992 66. Francis PJ, Berry V, Moore AT et al: Lens biology: Development and human cataractogenesis. Trends Gen 15:191, 1999 67. Kuck JFR Jr, Kuwabara T, Kuck KD: The Emory mouse cataract: An animal model for human senile cataract. Curr Eye Res 2:106, 1982 68. Kuck JFR: Late-onset hereditary cataract in the Emory mouse. A model for human senile
cataract. Exp Eye Res 50:659, 1990 69. Graw J: Cataract mutations and lens development. Prog Retinal Eye Res 18:235, 1999 70. Graw J: Mouse models of congenital cataract. Eye 13:438, 1999 71. Tumminia SJ, Jonak GJ, Focht RJ et al: Cataractogenesis in transgenic mice containing the HIV-1 protease linked
to the lens αA-crystallin promoter. J Biol Chem 271:425, 1996 72. Mitton KP, Kamiya T, Tumminia SJ et al: Cysteine protease activated by expression of HIV-1 protease in transgenic
mice. J Biol Chem 271:31803, 1996 73. Egwuagu CE, Sztein J, Chan C-C et al: Ectopic expression of gamma interferon in the eyes of transgenic mice induces
ocular pathology and MHC class II gene expression. Invest Ophthalmol Vis Sci 35:332, 1994 74. Bloemendal H, Benedetti EL, Dunia I: Transgenic mice: Models for the study of cataract. Ophthal Res 28(Suppl): 1, 1996 75. Cammarata PR, Zhou C, Chen G et al: A transgenic animal model of osmotic cataract. Part I: Overexpression of
bovine Na+ /myoinositol cotransporter in lens fibers. Invest Ophthalmol Vis Sci 40:1727, 1999 76. Brady JP, Garland D, Duglas-Tabor Y et al: Targeted disruption of the mouse αA-crystallin gene induces cataract
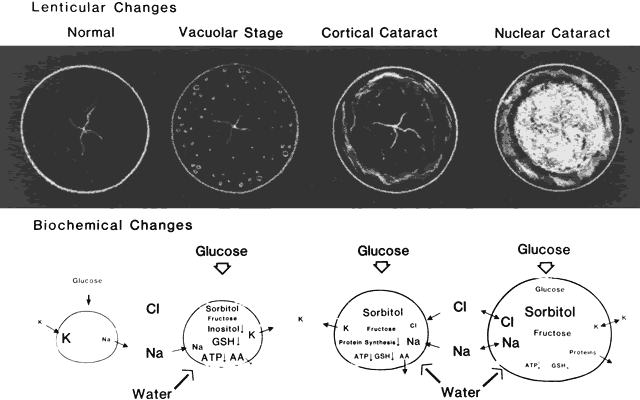
and cytoplasmic inclusion bodies containing the small heat shock protein αB-crystallin. Proc Natl Acad Sci USA 94:884, 1997 77. Wawrousek EF, Brady JP: αB-Crystallin gene knockout mice develop a severe, fatal phenotype
late in life. Invest Ophthal Vis Sci 39:S523, 1998 78. Gong GH, Agopian K, Kumar NM et al: Genetic factors influence cataract formation in alpha (3) connexin knockout
mice. Dev Genetics 24:27, 1999 79. Kador PF: Overview of the current attempts toward the medical treatment of cataract. Ophthalmology 90:352, 1983 80. Hockwin O, Schmitt C: Stellenwert der Antikataraktica. Fortschr Ophthalmol 87:9, 1990 81. Robison WG Jr, Laver NM: Ocular lesions in animal models of human diabetes. In
Shafrir E (ed): Lessons From Animal Diabetes IV, p 145. London: Smith-Gordon, 1993 82. Jedziniak JA, Chylack LT Jr, Cheng HM et al: The sorbitol pathway in the human lens: Aldose reductase and polyol dehydrogenase. Invest Ophthalmol Vis Sci 20:314, 1981 83. Lerner BC, Varma S, Richards R: Polyol pathway metabolites in human cataracts. Arch Ophthalmol 102:917, 1984 84. Ederer F, Hiller R, Taylor H: Senile lens changes and diabetes in two population studies. Am J Ophthalmol 91:381, 1981 85. Datiles MB III, Kador PF: Type I diabetic cataract. Arch Ophthalmol 117:284, 1999 86. Lou MF, Xu GT, Zigler JS Jr et al: Inhibition of naphthalene cataract in rats by aldose reductase inhibitors. Curr Eye Res 15:423, 1996 87. Sato S, Sugiyama K, Lee YS et al: Prevention of naphthalene-1,2-dihydrodiol-induced lens protein modifications
by structurally diverse aldose reductase inhibitors. Exp Eye Res 68: 601, 1999 88. Christen WG, Glynn RJ, Hennekens CH: Antioxidants and age-related eye disease: Current and future perspectives. Ann Epidemiol 6:60, 1996 89. Devamanoharan PS, Henein M, Morris S et al: Prevention of selenite cataract by vitamin C. Exp Eye Res 52:563, 1991 90. Vinson JA, Possanza CJ, Drack AV: The effect of ascorbic acid on galactose-induced cataracts. Nutr Rep Int 33:665, 1986 91. Stewart-Dehaan PJ, Dzialoszynski T, Trevithick JR: Modeling cortical cataractogenesis XXIV: Uptake by the lens of glutathione
injected into the rat. Mol Vis 5:37, 1999 92. Kassoff A, Kieval S, Mehu M et al: The Age-Related Eye Disease Study (AREDS): Design implications, AREDS report
No. 1. Control Clin Trials 20:573, 1999 93. Murray DL, Rathbun WB: Conditions for maximizing and inhibiting synthesis of glutathione in cultured
rat lenses: An application of HPLC with radioisotope detection. Curr Eye Res 9:55, 1990 94. Rathbun WB, Holleschau AM, Cohen JF et al: Prevention of acetaminophen- and naphthalene-induced cataract and glutathione
loss by CySSME. Invest Ophthalmol Vis Sci 37: 923, 1996 95. Sasaki H, Lin LR, Yokoyama T et al: TEMPOL protects against lens DNA strand breaks and cataract in the X-rayed
rabbit. Invest Ophthalmol Vis Sci 39:544, 1998 96. Bantseev V, Bhardwaj R, Rathbun W et al: Antioxidants and cataract: Cataract induction in space environment and
application to terrestrial aging cataract. Biochem Mol Biol Int 42:1189, 1997 97. Varma SD, Devamanoharan PS, Morris SM: Prevention of cataracts by nutritional and metabolic antioxidants. Crit Rev Food Sci Nutr 35:111, 1995 98. Cotlier E, Sharma YR: Aspirin and senile cataracts in rheumatoid arthritis. Lancet 1:338, 1981 99. Sperduto RD: Epidemiological aspects of age-related cataract. In Tasman
W, Jaeger EA (eds): Duane's Clinical Ophthalmology, Chapter 73A, p 4. Philadelphia: JB
Lippincott, 1997 |