|
|
Chapter 10: Retina
Authors:
Retina
I. RETINA
The human retina is a highly organized structure, consisting of alternate layers of cell bodies and synaptic processes. Despite its compact size and apparent simplicity when compared with nervous structures such as the cerebral cortex, the retina has a remarkably sophisticated level of processing power. Visual processing of the retina is elaborated upon by the brain, and the perception of color, contrast, depth, and form occurs in the cortex.
The anatomy of the retina is presented in Chapter 1. Figure 1-17 shows the major cell types and identifies the layers of this tissue. Division of the retina into layers composed of groups of similar cells permits the clinician to localize a function or functional disturbance to a single layer or group of cells. Processing of retinal information proceeds from the photoreceptor layer through the ganglion cell axon to the optic nerve and brain.
PHYSIOLOGY
The retina is the most complex of the ocular tissues. In order to see, the eye must perform as an optical instrument, as a complex receptor, and as an effective transducer. Rod and cone cells in the photoreceptor layer are capable of transforming light stimulus into a nerve impulse that is conducted by the nerve fiber layer of the retina through the optic nerve and ultimately to the occipital visual cortex. The macula is responsible for the best visual acuity and for color vision, and most of its photoreceptor cells are cones. In the central fovea, there is a nearly 1:1 relationship between the cone photoreceptor, its ganglion cell, and the emerging nerve fiber, and this ensures the most acute vision. In the peripheral retina, many photoreceptors are coupled to the same ganglion cell, and a more complex system of relays is necessary. The result of such an arrangement is that the macula is used primarily for central and color vision (photopic vision) while the remaining retina, which is populated mostly by rod photoreceptors, is utilized primarily for peripheral and night (scotopic) vision.
The rod and cone photoreceptors are located in the avascular outermost layer of the sensory retina and are the site of the chemical reaction initiating the visual process. Each rod photoreceptor cell contains rhodopsin, which is a photosensitive visual pigment formed when opsin protein molecules combine with 11-cis retinal. As a photon of light is absorbed by rhodopsin, 11-cis retinal is immediately isomerized to its all-trans form. Rhodopsin is a membrane-bound glycolipid that is partially embedded in the double membrane disks of the photoreceptor outer segment. Peak light absorption by rhodopsin occurs at approximately 500 nm, which is the blue-green region of the light spectrum. Spectral sensitivity studies of cone photopigments have shown peak wavelength absorption at 430, 540, and 575 nm for blue-, green-, and red-sensitive cones, respectively. The cone photopigments are composed of 11-cis retinal bound to a variety of opsin proteins.
Scotopic vision is mediated entirely by the rod photoreceptors. With this dark-adapted form of vision, varying shades of gray are seen, but colors cannot be distinguished. As the retina becomes fully light-adapted, the spectral sensitivity of the retina shifts from a rhodopsin-dominated peak of 500 nm to approximately 560 nm, and color sensation becomes evident. An object takes on color when it contains photopigments that absorb specific wavelengths and selectively reflect or transmit certain wavelengths of light within the visible spectrum (400-700 nm). Daylight vision is mediated primarily by cone photoreceptors, twilight by a combination of cones and rods, and night vision by the rod photoreceptors.
EXAMINATION
The examination of the retina is described in Chapter 2 and depicted in Figures 2-12, 2-13, 2-14, 2-15, 2-16, 2-17 and 2-18. The retina can be examined with a direct or indirect ophthalmoscope or with a slitlamp (biomicroscope) and contact or handheld biconvex lens. With these instruments, the skilled observer is clinically able to dissect the layers of the retina in order to determine the type, level, and extent of retinal disease. Fundus photography and fluorescein angiography (Figures 2-27, 2-28 and 2-29) are useful adjuncts to the clinical examination; photography allows pictorial documentation for future comparison, and angiography provides the vascular detail needed for laser treatment of retinal diseases.
The clinical application of visual electrophysiologic and psychophysical tests is described in Chapter 2. Such tests may be helpful in establishing the diagnosis of certain disease entities.
DISEASES OF THE MACULA
AGE-RELATED MACULAR DEGENERATION
Age-related macular degeneration is the leading cause of permanent blindness in the elderly. The exact cause is unknown, but the incidence increases with each decade over age 50. Other associations besides age include race (usually Caucasian), sex (slight female predominance), family history, and a history of cigarette smoking. The disease includes a broad spectrum of clinical and pathologic findings that can be classified into two groups: nonexudative ("dry") and exudative ("wet"). Although both types are progressive and usually bilateral, they differ in their manifestations, prognosis, and management. The more severe exudative form accounts for approximately 90% of all cases of legal blindness due to age-related macular degeneration.
1. NONEXUDATIVE MACULAR DEGENERATION
Nonexudative age-related macular degeneration is characterized by variable degrees of atrophy and degeneration of the outer retina, retinal pigment epithelium, Bruch's membrane and choriocapillaris. Of the ophthalmoscopically visible changes in the retinal pigment epithelium and Bruch's membrane, drusen are the most typical (Figure 10-1). Drusen are discrete, round, yellow-white deposits of variable size beneath the pigment epithelium and are scattered throughout the macula and posterior pole. With time, they may enlarge, coalesce, calcify, and increase in number. Histopathologically, most drusen consist of focal collections of eosinophilic material lying between the pigment epithelium and Bruch's membrane; they therefore represent focal detachment of the pigment epithelium. In addition to drusen, clumps of pigment irregularly dispersed within depigmented areas of atrophy may progressively appear throughout the macula. The level of associated visual impairment is variable and may be minimal. Fluorescein angiography demonstrates irregular patterns of retinal pigment epithelial hyperplasia and atrophy. Electrophysiologic testing in most patients is normal.
There is no generally accepted treatment or means of prevention of this type of macular degeneration. Laser retinal photocoagulation appears to have a beneficial effect on drusen but has not yet been shown to improve visual outcome. Although high plasma levels of antioxidants are associated with a reduced risk of age-related macular degeneration, the use of vitamin supplements does not appear to be preventive. Most patients with macular drusen never experience significant loss of central vision; the atrophic changes may stabilize or progress slowly. However, the exudative stage may develop suddenly at any time, and in addition to regular ophthalmic examinations, patients are given an Amsler grid (Figure 2-22) to help monitor and report any symptomatic changes.
2. EXUDATIVE MACULAR DEGENERATION
Although patients with age-related macular degeneration usually manifest nonexudative changes only, the majority of patients who experience severe vision loss from this disease do so from the development of subretinal neovascularization and related exudative maculopathy. Serous fluid from the underlying choroid can leak through small defects in Bruch's membrane, causing focal detachment of the pigment epithelium. Additional fluid may lead to further separation of the overlying sensory retina, and vision usually decreases if the fovea is involved. Retinal pigment epithelial detachments may spontaneously flatten, with variable visual results, and leave a geographic area of depigmentation at the involved site.
Ingrowth of new vessels from the choroid into the subretinal space is the most important change that predisposes patients with drusen to macular detachment and irreversible loss of central vision. These new vessels grow in a flat cartwheel or sea-fan configuration away from their site of entry into the subretinal space. The clinical changes of early subretinal neovascularization are subtle and may be easily overlooked; during this occult stage of new vessel formation, the patient is asymptomatic, and the new vessels may not be apparent either ophthalmoscopically or angiographically.
The ophthalmologist must maintain a high index of suspicion that subretinal neovascularization is present whenever a patient with evidence of age-related macular degeneration has sudden or recent central vision loss, including blurred vision, distortion, or a new scotoma. If the fundus examination reveals subretinal blood, exudate, or a grayish-green choroidal lesion in the macula, there is great likelihood that neovascularization is present, and a fluorescein or indocyanine green angiogram should be obtained promptly to determine if a treatable lesion can be identified.
Although some subretinal neovascular membranes may spontaneously regress, the natural course of subretinal neovascularization in age-related macular degeneration is toward irreversible loss of central vision over a variable period of time. The sensory retina may be damaged by long-standing edema, detachment, or underlying hemorrhage. Furthermore, a hemorrhagic detachment of the retina may undergo fibrous metaplasia, resulting in an elevated subretinal mass called a disciform scar. This elevated fibrovascular mound of variable size represents the cicatricial end stage of exudative age-related macular degeneration. It is usually centrally located and results in permanent loss of central vision.
Treatment
In the absence of subretinal neovascularization, no medical or surgical treatment of serous retinal pigment epithelial detachment is of proved benefit. The use of parenteral alpha interferon, for example, has not been effective for this disease. However, if a well-defined extrafoveal (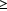 200
200 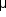 m from the center of the foveal avascular zone) subretinal neovascular membrane is present, laser photocoagulation is indicated. Angiography defines the precise location and borders of the neovascular membrane, which is then completely ablated by heavy confluent laser burns. Photocoagulation destroys the overlying retina as well but is worthwhile if the subretinal membrane can be halted short of the fovea (see Chapter 24).
m from the center of the foveal avascular zone) subretinal neovascular membrane is present, laser photocoagulation is indicated. Angiography defines the precise location and borders of the neovascular membrane, which is then completely ablated by heavy confluent laser burns. Photocoagulation destroys the overlying retina as well but is worthwhile if the subretinal membrane can be halted short of the fovea (see Chapter 24).
Krypton laser photocoagulation of juxtafoveal (<200 m from the center of the foveal avascular zone) subretinal neovascularization is recommended in nonhypertensive patients. The Macular Photocoagulation Study Group has refined its treatment recommendations for subfoveal disease and shown that selected patients may benefit from laser photocoag-ulation. The ability to determine the probable rate and direction of growth of a subretinal neovascular membrane would facilitate clinical decisions about if and when to treat a given membrane in cases where treatment indications are unclear. Unfortunately, many patients with exudative macular degeneration present with subretinal neovascularization that is either not sufficiently well defined or is too extensive for laser photocoagulation to be useful.
Following successful photocoagulation of a subretinal neovascular membrane, recurrent neovascularization either contiguous with or remote from the laser scar may occur in one-half of cases by 2 years. Recurrence is often accompanied by severe vision loss, so that careful monitoring with Amsler grids, ophthalmoscopy, and angiography is essential. Low-dose radiotherapy has provided encouraging results in patients with subfoveal neovascularization. Patients with impaired central vision in both eyes may benefit from a variety of low vision aids.
CENTRAL SEROUS CHORIORETINOPATHY
Central serous chorioretinopathy is characterized by serous detachment of the sensory retina as a consequence of focal leakage of fluid from the choriocapillaris through a defect in the retinal pigment epithelium (Figures 10-2 and 10-3). This disease typically affects young to middle-aged men and may be related to life stress events. Most patients present with the sudden onset of blurred vision, micropsia, metamorphopsia, and central scotoma. Visual acuity is often only moderately decreased and may be improved to near-normal with a small hyperopic correction.
The diagnosis is made by slitlamp examination of the fundus; the presence of serous detachment of the sensory retina in the absence of ocular inflammation, subretinal neovascularization, an optic pit, or a choroidal tumor is diagnostic. The retinal pigment epithelial lesion appears as a small, round or oval, yellowish-gray spot that is variable in size and may be difficult to detect without the aid of fluorescein angiography. Fluorescein dye leaking from the choriocapillaris may accumulate below the pigment epithelium or sensory retina, resulting in a variety of patterns including the well-recognized smokestack configuration.
Approximately 80% of eyes with central serous chorioretinopathy undergo spontaneous resorption of subretinal fluid and recovery of normal visual acuity within 6 months after the onset of symptoms. Despite normal acuity, however, many patients have a mild permanent visual defect, such as a decrease in color sensitivity, micropsia, or relative scotoma. Twenty to 30 percent of patients will have one or more recurrences of the disease, and complications-including subretinal neovascularization and chronic cystoid macular edema-have been described in patients with frequent and prolonged serous detachments.
The cause of central serous chorioretinopathy is unknown; there is no convincing evidence that the disease is either infectious or due to retinal pigment epithelial dystrophy. Argon laser photocoagulation directed to the active leak significantly shortens the duration of the sensory detachment and hastens the recovery of central vision, but there is no evidence that prompt photocoagulation reduces the chance of permanent loss of visual function. Although the complications of retinal laser photocoagulation are few, it is probably not advisable to recommend immediate photocoagulation treatment in all patients with central serous chorioretinopathy. The duration and location of disease, the condition of the fellow eye, and occupational visual requirements are all considerations upon which treatment decisions are based.
MACULAR EDEMA
Retinal edema involving the macula may be associated with a variety of intraocular inflammatory diseases, retinal vascular diseases, intraocular surgery, inherited or acquired retinal degenerations, medications, macular membranes, or unknown causes. Macular edema may be diffuse, with nonlocalized intraretinal fluid causing thickening of the macula. When edema fluid accumulates in honeycomb-like spaces of the outer plexiform and inner nuclear layers, it is called cystoid macular edema. On fluorescein angiography, fluorescein dye leaks from the perifoveal retinal capillaries and accumulates in a flower-petal pattern about the fovea (Figure 10-4).
The most widely recognized association with cystoid macular edema is intraocular surgery. Approximately 50% of eyes undergoing uneventful intracapsular cataract extraction and 20% of eyes undergoing extracapsular cataract extraction develop angiographic cystoid macular edema. Clinically significant edema usually occurs within 4-12 weeks postoperatively, but in some instances its onset may be delayed for months or years. Many patients with cystoid macular edema of less than 6 months' duration have self-limited leakage that will resolve without treatment. Topical or local (or both) anti-inflammatory therapy may be of value in restoring visual acuity in some patients with chronic postoperative macular edema. YAG laser vitreolysis (see Chapter 24) and surgical vitrectomy may be of benefit when the macular edema is associated with vitreous tissue incarcerated in the cataract wound or adherent to anterior segment structures. When an intraocular lens implant is the cause of postoperative macular edema due to its design, positioning, or inadequate fixation, removal of the lens implant can be considered.
INFLAMMATORY DISORDERS INVOLVING THE MACULA
Presumed Ocular Histoplasmosis Syndrome (Figures 10-5, 10-6 and 10-7)
In this disease, serous and hemorrhagic detachments of the macula are associated with multiple peripheral atrophic chorioretinal scars and peripapillary chorioretinal scarring (see Chapter 7). The syndrome usually occurs in healthy patients between the third and sixth decades of life, and the scars are probably caused by an antecedent subclinical systemic infection with Histoplasma capsulatum. The macular detachments are due to subretinal neovascularization, and the visual prognosis depends on the proximity of the neovascular membrane to the center of the fovea. If the membrane extends inside the foveal avascular zone, only 15% of eyes will retain 20/40 vision. A macular scar may change over time, and 10% of patients with normal maculae will develop new atrophic scars in this region. The relative risk of developing macular subretinal neovascularization in the second eye of an affected patient is significant, and these patients should be instructed in the frequent use of the Amsler grid and the importance of prompt examination when changes are detected.
Argon laser photocoagulation of a subretinal neovascular membrane outside the foveal avascular zone in symptomatic patients is of value in preventing severe vision loss. The surgical removal of submacular membranes may prove useful in preserving vision.
Acute Multifocal Posterior Placoid Pigment Epitheliopathy (AMPPPE)
AMPPPE typically affects healthy young patients who develop rapidly progressive bilateral vision loss in association with ophthalmoscopically visible multifocal flat gray-white subretinal lesions involving the pigment epithelium (Figure 10-8). The cause of this disease, which in many instances is associated with evidence of an influenza-like illness, is unknown; the course and nature of the illness suggests the possibility of viral infection. The characteristic feature of the disease is the rapid resolution of the fundus lesions and a delayed return of visual acuity to near-normal levels. Although the prognosis for visual recovery in this acute self-limited disease is good, many patients will identify small residual paracentral scotomas when carefully tested. Extensive pigmentary changes remaining during the late stages of AMPPPE may mimic widespread retinal degeneration; the clinical history and normal electrophysiologic findings aid in this differential diagnosis.
Geographic Helicoid Peripapillary Choroidopathy
This is a chronic progressive and recurrent multifocal inflammatory disease of the retinal pigment epithelium, choriocapillaris, and choroid. It characteristically involves the juxtapapillary retina and extends radially to involve the macula and peripheral retina. The active stage manifests itself as sharply demarcated gray-yellow lesions with irregular borders that appear to involve the pigment epithelium and choriocapillaris. Vitritis, anterior uveitis, and subretinal neovascularization have been associated with this disorder. Involvement is usually bilateral, and the cause is unknown. The natural history of this indolent inflammatory disease is variable and may correlate with the presence of disease in the fellow eye. Local or systemic corticosteroid treatment may be of benefit when active inflammation is present; laser photocoagulation is administered as indicated for the complication of subretinal neovascularization.
Vitiliginous Chorioretinitis (Birdshot Retinochoroidopathy)
This is a syndrome characterized by diffuse cream-colored patches at the level of the pigment epithelium and choroid, retinal vasculitis associated with cystoid macular edema, and vitritis. The associations with HLA-A29 and with retinal S-antigen suggest that this disease has a genetic predisposition and that retinal autoimmunity plays a role in its manifestations. In many cases, electroretinography, electro-oculography, and dark adaptation studies are abnormal. The course of the disease is that of exacerbation and remission with variable visual outcomes; visual loss has been attributed to chronic cystoid macular edema, optic atrophy, macular scarring, or subretinal neovascularization. Corticosteroid therapy has not proved effective against this disease.
Acute Macular Neuroretinopathy
Acute macular neuroretinopathy is characterized by the acute onset of paracentral scotomas and mild visual acuity loss accompanied by wedge-shaped parafoveal retinal lesions in the deep sensory retina of one or both eyes. The macular lesions are subtle, reddish-brown, and best seen with a red-free light. The patients are usually young adults with a history of acute viral illness. While the retinal lesions may fade, the scotomas tend to persist and remain symptomatic.
Multiple Evanescent White Dot Syndrome
This is an acute and self-limited unilateral disease that affects mainly young women and is characterized clinically by multiple white dots at the level of the pigment epithelium, vitreal cells, and transient electroretinographic abnormalities. The cause is unknown. There is no evidence of associated systemic disease. The retinal lesions gradually regress in a matter of weeks, leaving only minor retinal pigment epithelial defects.
ANGIOID STREAKS
Angioid streaks appear as irregular, jagged tapering lines that radiate from the peripapillary retina into the macula and peripheral fundus (Figure 10-9). The streaks represent linear crack-like dehiscences in Bruch's membrane. The lesions are rarely noted in children and probably develop in the second or third decade of life. Early in the disease the streaks are sharply outlined and red-orange or brown. Subsequent fibrovascular tissue growth may partially or totally obscure the streak margins.
Nearly 50% of patients with angioid streaks have an associated systemic disease. Pseudoxanthoma elasticum, Paget's disease of bone, Ehlers-Danlos syndrome, and several hemoglobinopathies and hemolytic disorders have been associated with this retinal disease, but the most common association is with age-related degeneration of Bruch's membrane. Patients with angioid streaks should be warned of the potential risk of choroidal rupture from even relatively mild eye trauma. Older patients with the disease are at risk of developing serous and hemorrhagic detachments of the retina as a consequence of subretinal neovascularization.
Laser treatment may be used to photocoagulate extrafoveal neovascular membranes; however, other neovascular membranes are likely to occur. Prophylactic treatment of angioid streaks before subretinal neovascularization develops is not recommended.
MYOPIC MACULAR DEGENERATION
Pathologic myopia is one of the leading causes of blindness in the United States and is characterized by progressive elongation of the eye with subsequent thinning and atrophy of the choroid and pigment epithelium in the macula. Peripapillary chorioretinal atrophy and linear breaks in Bruch's membrane ("lacquer cracks") are characteristic findings on ophthalmoscopy (Figure 10-10). The degenerative changes of the macular pigment epithelium resemble those found in the older patient with age-related macular degeneration. A characteristic lesion of this disease is a raised, circular, pigmented macular lesion called a Fuchs spot. Most patients are in the fifth decade when the degenerative macular changes cause a slowly progressive loss of vision; rapid loss of visual acuity is usually caused by serous and hemorrhagic macular degeneration overlying a subretinal neovascular membrane.
Fluorescein angiography in patients with pathologic myopia may show delayed filling of choroidal and retinal blood vessels. Angiography is helpful in identifying and locating the site of subretinal neovascularization in patients who develop serous or hemorrhagic detachments of the macula. Because of the frequent close proximity of the subretinal neovascular membrane to the foveola in these patients, laser photocoagulation may not be possible. As subretinal neovascular membranes tend to remain small and because photocoagulation-associated chorioretinal atrophy tends to progress in patients with pathologic myopia, retinal laser treatment is not as beneficial as in other diseases associated with macular subretinal neovascularization.
The chorioretinal changes of pathologic myopia predispose the retina to breaks and thus to retinal detachment. Peripheral retinal findings may include paving stone degeneration, pigmentary degeneration, and lattice degeneration. Retinal breaks usually occur in areas involved with chorioretinal lesions, but they also arise in areas of apparently normal retina. Some of these breaks, particularly those of the "horseshoe" and round retinal tear type, will progress to rhegmatogenous retinal detachment.
MACULAR HOLE
A macular hole is a partial or full-thickness absence of the sensory retina in the macula. This disorder occurs most often in elderly women and is associated with elevated plasma fibrinogen levels. The typical finding on biomicroscopy of the symptomatic eye is a full-thickness, round or oval, sharply defined hole measuring one-third disk diameter in the center of the macula, which may be surrounded by a ring detachment of the sensory retina (Figure 10-11). With a full-thickness macular hole, visual acuity is impaired and metamorphopsia, as well as a central scotoma, are present on the Amsler grid. An operculum of retinal tissue may overlie the macular hole. Tangential traction from epiretinal vitreous cortex plays an important role in the pathogenesis of macular hole. Early stages of macular hole formation, such as a deep foveal yellow spot or ring, may be reversible as the posterior vitreous cortex spontaneously separates from the retina. Therapy for macular hole disease involves reattaching and potentially restoring function to the retina overlying the cuff of subretinal fluid surrounding the hole. While the anatomic results of vitrectomy surgery to close macular holes are encouraging, the clinical benefits are still under study.
EPIRETINAL MACULAR MEMBRANES
Fibrocellular membranes may proliferate on the surface of the retina, either in the macula or peripheral retina. Contraction or shrinkage of these epiretinal membranes may cause varying degrees of visual distortion, intraretinal edema, and degeneration of the underlying retina. Biomicroscopy usually shows retinal wrinkles and vessel tortuosity and may rarely also show retinal hemorrhages, cotton-wool spots, serous retinal detachment, and macular hole; a posterior vitreous detachment is nearly always present (Figure 10-12). Disorders associated with epiretinal membranes include retinal tears with or without rhegmatogenous retinal detachment, vitreous inflammatory diseases, trauma, and a variety of retinal vascular diseases.
Patients with macular distortion and vision loss caused by epiretinal membrane contraction are usually left with stable visual acuity, suggesting that membrane contraction is a short-lived and self-limited process. Surgical peeling of severe epiretinal membranes can be performed successfully, but regrowth of epiretinal tissues occurs in some cases. There is no role for photocoagulation in the treatment of epiretinal macular membrane disease.
TRAUMATIC MACULOPATHY
Blunt trauma to the anterior segment of the eye may cause a contrecoup injury to the retina called commotio retinae. The retina develops a gray-white color that affects primarily the outer retina and may be confined to the macular area (Berlin's edema) or may involve extensive areas of the peripheral retina. The retinal whitening in the macular area may clear completely, or impairment of central vision may be permanent and associated with a pigmented retinal scar (Figure 10-13) or a macular hole. Trauma similar to that which causes Berlin's edema may also cause choroidal rupture with subretinal hemorrhage and permanent central vision loss.
In addition to blunt trauma, several other traumatic injuries involving the macula are of importance. Purtscher's retinopathy is characterized by multiple patches of superficial retinal whitening and retinal hemorrhages in each eye of a patient after severe compression injury to the head or trunk. Terson's syndrome is seen in approximately 20% of patients after traumatic (or spontaneous) subarachnoid or subdural hemorrhage and is characterized by vitreous and superficial macular hemorrhage. Solar retinopathy refers to a specific foveolar lesion that occurs after sun-gazing and is best described as a usually bilateral sharply circumscribed and often irregularly shaped partial-thickness hole or depression in the center of the fovea.
MACULAR DYSTROPHIES
Macular dystrophies differ from degenerations in that the former are inherited, though not necessarily evident at birth, and are not associated with systemic diseases. Most often the disorder is restricted to the macula; it may be symmetric or asymmetric, but eventually both eyes are affected. In the early stages of some of these disorders the visual acuity may be reduced while the macular changes are subtle or absent on ophthalmoscopy, and the patient's complaint may be dismissed as spurious. Conversely, in other macular dystrophies, the ophthalmoscopic changes may be very striking at a time when the patient is free of visual symptoms. One method of classifying the more common macular dystrophies is to consider the presumptive anatomic layer or layers of the retina involved (Table 10-1).
X-Linked Juvenile Retinoschisis
This is a congenital disease of males characterized by a macular lesion called "foveal schisis." On slitlamp examination, foveal schisis appears as small superficial retinal cysts arranged in a stellate pattern accompanied by radial striae centered in the foveal area (Figure 10-14). Visual acuity is usually between 20/40 and 20/200; peripheral visual field abnormalities are present in the 50% of patients with associated peripheral retinoschisis. The posterior pole appears normal on fluorescein angiography, and this may be helpful in the clinical differentiation from cystoid macular edema. B wave abnormalities on the electroretinogram are consistent with the histopathologic finding of intraretinal splitting in the nerve fiber layer.
Cone-Rod Dystrophies
The cone-rod dystrophies constitute a relatively rare group of disorders that may be regarded as a single entity showing variable expressivity. Most cases are sporadic, but familial cases are usually transmitted by an autosomal dominant inheritance pattern. Cone-rod dystrophy is characterized by predominant involvement of the cone photoreceptors with progressive color vision defects and associated loss of visual acuity. A bilateral and symmetric bulls-eye pattern of depigmentation and a corresponding zone of hyperfluorescence surrounding a central nonfluorescent spot (similar to that seen in chloroquine retinopathy) are the most commonly described biomicroscopic and angiographic changes in these patients (Figure 10-15). As the disease progresses, the electroretinogram shows marked loss of cone function associated with a slight to moderate loss of rod function. Histopathologic study shows absence of macular and paramacular photoreceptors, and there is associated pigment epithelium degeneration.
Fundus Albipunctatus
Fundus albipunctatus is an autosomal recessive nonprogressive dystrophy characterized by a myriad of discrete small white dots at the level of the pigment epithelium sprinkled about the posterior pole and midperiphery of the retina. Patients are night-blind with normal visual acuity, normal visual fields, and normal color vision. While the electroretinogram and electro-oculogram are usually normal, dark adaptation thresholds are markedly elevated. Retinitis punctata albescens is the less common progressive variant of this dystrophy.
Fundus Flavimaculatus (Stargardt's Disease)
This is a bilateral and symmetric autosomal recessive disorder characterized by multiple yellow-white fleck lesions of variable size and shape confined to the retinal pigment epithelium (Figure 10-16). Many patients suffer central visual loss in childhood; however, macular involvement and the ultimate visual outcome are variable. Fluorescein angiography is important in differentiating flecks from drusen; the former are usually hypofluorescent. The electroretinogram and electro-oculogram are usually normal. Histopathologic abnormalities are confined to the pigment epithelium; the yellow flecks seen clinically are dense accumulations of lipofuscin within engorged pigment epithelial cells.
Vitelliform Dystrophy (Best's Disease)
Vitelliform dystrophy is an autosomal dominant disorder with variable penetrance and expressivity with onset usually in childhood. The ophthalmoscopic appearance is variable and ranges from a mild pigmentary disturbance within the fovea to the typical vitelliform or "egg yoke" lesion located within the central macula (Figure 10-17). This characteristic cyst-like lesion is generally quite round and well demarcated and contains homogeneous opaque yellow material lying at the apparent level of the retinal pigment epithelium. The "egg yoke" may degenerate and be associated with subretinal neovascularization, subretinal hemorrhage, and extensive macular scarring. Visual acuity often remains good, and the electroretinogram is normal; the distinctly abnormal electro-oculogram is the hallmark of this disease.
DISEASES OF THE PERIPHERAL RETINA
RETINAL DETACHMENT
The term "retinal detachment" denotes separation of the sensory retina, ie, the photoreceptors and inner tissue layers, from the underlying retinal pigment epithelium. There are three main types: rhegmatogenous detachment, traction detachment, and serous or hemorrhagic detachment.
1. RHEGMATOGENOUS RETINAL DETACHMENT
The most common of the three major types of retinal detachments is rhegmatogenous retinal detachment. The characteristics of a rhegmatogenous detachment are a full-thickness break (a "rhegma") in the sensory retina, variable degrees of vitreous traction, and passage of liquefied vitreous through the sensory retinal defect into the subretinal space. A spontaneous rhegmatogenous retinal detachment is usually preceded or accompanied by a posterior vitreous detachment. Myopia, aphakia, lattice degeneration, and ocular trauma are associated with this type of retinal detachment. Binocular indirect ophthalmoscopy with scleral depression (Figures 2-16 and 2-18) reveals elevation of the translucent detached sensory retina. A careful search usually reveals one or more full-thickness sensory retinal breaks such as a horseshoe tear, round atrophic hole, or anterior circumferential tear (retinal dialysis). The location of retinal breaks varies according to type; horseshoe tears are most common in the superotemporal quadrant, atrophic holes in the temporal quadrants, and retinal dialysis in the inferotemporal quadrant. When multiple retinal breaks are present, the defects are usually within 90 degrees of one another.
Treatment
Scleral buckling or pneumatic retinopexy are the two most popular and effective surgical techniques for the repair of rhegmatogenous retinal detachment. Each procedure requires careful localization of the retinal break and treatment with cryotherapy or laser in order to create an adhesion between the pigment epithelium and the sensory retina. With scleral buckling surgery, the retinal break is mounted on sclera indented by an explant. The scleral indentation can be achieved by a variety of techniques and materials, each of which has inherent advantages and disadvantages. Pneumatic retinopexy involves the intraocular injection of air or an expandable gas in order to tamponade the retinal break while the chorioretinal adhesion forms. An overall reattachment rate of 90% is reported; however, the visual results are dependent on the preoperative status of the macula. If the macula is involved in rhegmatogenous retinal detachment, the likelihood of complete visual recovery is slight.
2. TRACTION RETINAL DETACHMENT
Traction retinal detachment is the second most common type and is most commonly due to proliferative diabetic retinopathy, proliferative vitreoretinopathy, retinopathy of prematurity, or ocular trauma. In contrast to the convex appearance of rhegmatogenous retinal detachment, the typical traction retinal detachment has a more concave surface and is likely to be more localized, usually not extending to the ora serrata. The tractional forces that actively pull the sensory retina away from the underlying pigment epithelium are caused by a clinically apparent vitreal, epiretinal, or subretinal membrane consisting of fibroblasts and of glial and retinal pigment epithelial cells. In diabetic traction retinal detachment, vitreous contraction draws the fibrovascular tissue and underlying retina anteriorly toward the vitreous base. Initially the detachment may be localized along the vascular arcades, but progression may spread to involve the midperipheral retina and the macula. Proliferative vitreoretinopathy is a complication of rhegmatogenous retinal detachment and is the most common cause of failure of surgical repair in these eyes.
The basic pathologic process in eyes with proliferative vitreoretinopathy is growth and contraction of cellular membranes on both sides of the retina and on the posterior vitreous surface. Focal traction from cellular membranes can produce a retinal tear and lead to combined tractional-rhegmatogenous retinal detachment.
Treatment
The primary treatment of traction retinal detachment is vitreoretinal surgery and may involve vitrectomy, membrane removal, scleral buckling, and injection of intraocular gas or silicone oil.
3. SEROUS & HEMORRHAGIC RETINAL DETACHMENT
Serous and hemorrhagic retinal detachment can occur in the absence of either retinal break or vitreoretinal traction. These detachments are the result of a collection of fluid beneath the sensory retina and are caused primarily by diseases of the retinal pigment epithelium and choroid. Degenerative, inflammatory, and infectious diseases limited to the macula, including the multiple causes of subretinal neovascularization, may be associated with this third type of retinal detachment and are described in an earlier section of this chapter. This type of detachment may also be associated with systemic vascular and inflammatory disease as described in Chapters 7 and 15.
RETINOPATHY OF PREMATURITY
Retinopathy of prematurity is a vasoproliferative retinopathy that is the leading cause of childhood blindness in the United States and a major cause of blindness throughout the developed world. An international classification of this disease divides the retina into three zones and characterizes the extent of disease by the number of clock hours involved; the retinal changes are divided into five stages described in Table 10-2.
The demarcation line is a narrow white band that marks the junction of vascular and avascular retina in stage 1; it is the first definite ophthalmoscopic sign of retinopathy of prematurity. As this band increases in height, width, and volume and rises up from the plane of the retina, the ridge of stage 2 is seen. Neovascular proliferation along the posterior aspect of the ridge and extending into the vitreous defines stage 3. Stage 4 is characterized by subtotal retinal detachment, and the clinical sign of stage 5 is a funnel-shaped total retinal detachment.
Treatment
The treatment of retinopathy of prematurity is based on the classification and stage of the disease. It is important to note that a significant number of patients with retinopathy of prematurity undergo spontaneous regression. Peripheral retinal changes of regressed retinopathy of prematurity include avascular retina, peripheral folds, and retinal breaks; associated changes in the posterior pole may include straightening of the temporal vessels, temporal stretching of the macula, and retinal tissue that appears to be dragged over the disk (Figure 10-18). Other ocular findings of regressed retinopathy of prematurity include myopia (which may be asymmetric), strabismus, cataract, and angle-closure glaucoma.
While stage 1 and stage 2 disease require nothing more than observation, transscleral cryotherapy or laser photocoagulation to the avascular retina should be considered in eyes with stage 3 disease. Vitreoretinal surgery as described above in the section on traction retinal detachment may be appropriate for eyes with stage 4 or stage 5 disease. The etiology and treatment of retinopathy of prematurity as well as the recommended screening protocols are discussed in Chapter 17.
RETINAL DEGENERATIONS
This group of disorders encompasses a number of diseases with various ocular and, in some instances, systemic manifestations. In this section, several specific disorders will be used as prototypes with which to understand the major characteristics of retinal degenerations.
Retinitis Pigmentosa
Retinitis pigmentosa is a group of hereditary retinal degenerations characterized by progressive dysfunction of the photoreceptors and associated with progressive cell loss and eventual atrophy of several retinal layers. The typical form of this disease can be inherited as an autosomal recessive, autosomal dominant, or X-linked recessive trait; one-third of cases will have a negative family history. The hallmark symptoms of retinitis pigmentosa are night blindness (nyctalopia) and gradually progressive peripheral visual field loss. The most characteristic ophthalmoscopic findings are narrowing of the retinal arterioles, mottling of the retinal pigment epithelium, and peripheral retinal pigment clumping, referred to as "bone-spicule formation" (Figure 10-19). While retinitis pigmentosa is a generalized photoreceptor disorder, in most cases rod function is more severely affected, leading to subjective sensations associated with poor scotopic function. The electroretinogram usually shows either markedly reduced or absent retinal function; the electro-oculogram lacks the usual light rise. The fundus appearance of retinitis pigmentosa may be mimicked by several disorders, including chorioretinitis, trauma, vascular occlusion, and resolved retinal detachment.
The effects of supplemental vitamins on the progression of retinitis pigmentosa require further study before treatment recommendations can be made. Patients with the disease benefit from genetic counseling and appropriate referral to agencies that provide services to the visually impaired.
Leber's Congenital Amaurosis
Leber's congenital amaurosis is a group of disorders characterized by severe visual impairment or blindness from infancy with no discernible cause. The disorders are usually inherited in an autosomal recessive manner and may be associated with mental retardation, seizures, and renal or muscular abnormalities. The ophthalmoscopic findings are variable; most patients show either a normal fundus appearance or only subtle retinal pigment epithelial granularity and mild vessel attenuation. A markedly reduced or absent electroretinogram indicates generalized photoreceptor dysfunction, and in infants this test is the only method by which an absolute diagnosis can be made.
Gyrate Atrophy
Gyrate atrophy is an autosomal recessive disorder caused by reduced activity of ornithine aminotransferase, a mitochondrial matrix enzyme that catalyzes several amino acid pathways. The incidence of this disorder is relatively high in Finland, and the ophthalmologic features are the most prominent manifestations of the disease. Patients usually develop nyctalopia within the first decade of life, and progressive peripheral visual field loss follows. Characteristic sharply demarcated circular areas of chorioretinal atrophy develop in the midperiphery of the fundus during the teenage years and become confluent with macular involvement late in the course of the disease. The electroretinogram is decreased or absent, and the electro-oculogram is reduced.
Treatment approaches to this disease have included pyridoxine supplementation, restriction of dietary arginine, and supplemental dietary lysine.
Peripheral Chorioretinal Atrophy
Peripheral chorioretinal atrophy (paving stone degeneration) is a common chorioretinal degeneration found in nearly one-third of adult eyes. Ophthalmoscopically, the lesions appear as isolated or grouped, small, discrete, yellow-white areas with prominent underlying choroidal vessels and pigmented borders. Choroidal vascular insufficiency is thought to be the cause of this benign disorder because the pathologic changes are limited to that portion of the retina supplied by the choriocapillaris. Paving stone degeneration is not of great pathologic significance, though it may be a sign of peripheral vascular disease.
Lattice Degeneration
Lattice degeneration is the most common of the inherited vitreoretinal degenerations, with an estimated incidence of 7% of the general population. Lattice degeneration is more commonly found in myopic eyes and is frequently associated with retinal detachment, occurring in nearly one-third of retinal detachment patients. The ophthalmoscopic appearance may be that of localized round, oval, or linear retinal thinning, with pigmentation, branching white lines, and whitish-yellow flecks; the hallmarks of the disease are the thinned retina punctuated by sharp borders with firm vitreoretinal adhesions at the margins. The mere presence of lattice degeneration is not cause enough for prophylactic therapy. A strong family history of retinal detachments, retinal detachment in the fellow eye, high myopia, and aphakia are risk factors for retinal detachment in eyes with lattice degeneration, and prophylactic treatment with cryosurgery or laser photocoagulation may be warranted.
RETINOSCHISIS
Degenerative retinoschisis, unlike X-linked juvenile retinoschisis described above, is a common acquired peripheral retinal disorder that is believed to develop from preexisting peripheral cystoid degeneration. The cystic changes of peripheral cystoid degeneration are seen to some degree in virtually all adults. This cystoid degeneration is characterized by intraretinal microcysts that often coalesce, giving the appearance of lobulated, irregularly branching, tortuous channels. Peripheral cystoid degeneration may develop into either of two degenerative forms of retinoschisis, each of which is characterized by sharply demarcated and absolute visual field defects.
Typical degenerative retinoschisis occurs in 1% of adults and is a bilateral disease in one-third of affected patients. On clinical examination, the disorder appears as a round or ovoid area of retinal splitting with fusiform elevation of the inner layer and an optically empty schisis cavity. The retinal splitting occurs at the outer plexiform layer. Complications such as hole formation and marked posterior extension are very uncommon and rarely require treatment.
Reticular degenerative retinoschisis is characterized by round or oval areas of retinal splitting in which a bullous elevation of an extremely thin inner layer occurs, most commonly in the lower temporal quadrant. In this form of the disease, the splitting usually occurs in the nerve fiber layer, and typical peripheral cystoid degeneration is usually present anterior to the lesion. When retinal breaks are present in both the inner and the outer layers, progressive rhegmatogenous retinal detachment may develop and threaten the macula, thus requiring treatment.
RETINAL VASCULAR DISEASES
DIABETIC RETINOPATHY
Diabetic retinopathy is one of the leading causes of blindness in the Western world. The view that chronic hyperglycemia of diabetes mellitus is the major determinant of diabetic retinopathy is supported by the observation that retinopathy in young people with type I (insulin-dependent) diabetes does not occur for at least 3-5 years after the onset of this systemic disease. Similar results have been obtained for type II (non-insulin-dependent) diabetes, but in such patients the time of onset and therefore the duration of disease are more difficult to determine precisely. It is recommended that patients with type I diabetes mellitus be referred for ophthalmologic examination within 3 years after diagnosis and reexamined on at least an annual basis. Type II diabetic patients should be referred for ophthalmologic examination at the time of diagnosis and reexamined at least annually. As diabetic retinopathy can become particularly aggressive during pregnancy, any diabetic woman who becomes pregnant should be examined by an ophthalmologist in the first trimester and at least every 3 months thereafter until parturition.
In terms of both prognosis and treatment, it is useful to divide diabetic retinopathy into nonproliferative and proliferative categories. The prevalence of proliferative retinopathy in type I diabetics with 15 years of systemic disease is 50%. While the prevalence of proliferative disease at 15 years is much less in type II diabetics, the prevalence of macular edema as a function of the duration of systemic disease is the same in both groups.
1. NONPROLIFERATIVE DIABETIC RETINOPATHY
Diabetic retinopathy is a progressive microangiopathy characterized by small vessel damage and occlusion. The earliest pathologic changes are thickening of the capillary endothelial basement membrane and reduction of the number of pericytes. Background diabetic retinopathy is a clinical reflection of the hyperpermeability and incompetence of involved vessels. The capillaries develop tiny dot-like outpouchings called microaneurysms, while the retinal veins become dilated and tortuous (Figure 10-20).
Multiple hemorrhages may appear throughout different levels of the retina. Flame-shaped hemorrhages are so shaped because of their location within the horizontally oriented nerve fiber layer, while dot and blot hemorrhages are in the deeper retina, where cells and axons are vertically oriented.
Macular edema is the most frequent cause of visual loss among patients with background diabetic retinopathy. The edema is caused primarily by a breakdown of the inner blood-retinal barrier at the level of the retinal capillary endothelium, allowing leakage of fluid and plasma constituents into the surrounding retina. The edema may be focal or diffuse and appears clinically as thickened, cloudy retina with associated microaneurysms and intraretinal exudate. Circinate zones of yellow, lipid-rich exudate may form around clusters of microaneurysms and are most frequently centered in the temporal portion of the macula. While the prevalence of macular edema is 10% in the diabetic population as a whole, there is a dramatic increase in prevalence in eyes with more severe retinopathy.
With progressive microvascular occlusion, signs of increasing ischemia may be superimposed on the picture of background retinopathy and produce the clinical picture of preproliferative diabetic retinopathy. The most typical findings here are multiple cotton-wool spots, beading of the retinal veins, and irregular segmental dilation of the retinal capillary bed (intraretinal microvascular abnormalities). Closure of retinal capillaries surrounding the foveal avascular zone may cause significant ischemia, manifest clinically by the presence of large dark retinal hemorrhages and small thread-like macular arterioles. Eyes with macular edema and significant ischemia have a poorer visual prognosis-with or without laser treatment-than eyes with edema and relatively good perfusion.
The visual and electrophysiologic dysfunctions associated with diabetes probably result from the local vascular abnormalities and the systemic metabolic effects of the disease to which the retina is subjected. A characteristic blue-yellow color vision abnormality develops, and hue discrimination may be impaired. Contrast sensitivity may be reduced in patients, even in the presence of normal visual acuity. Visual field testing may show relative scotomas corresponding to areas of retinal edema and nonperfusion, and abnormalities in dark adaptation have also been described. Electroretinographic abnormalities bear a relationship to the severity of retinopathy and may aid in predicting progression of retinopathy. Fluorescein angiography is invaluable in defining the microvascular abnormalities of diabetic retinopathy (Figures 10-21 and 10-22). Large filling defects of capillary beds-"capillary nonperfusion"-show the extent of retinal ischemia (Figure 10-23) and are usually most prominent in the midperiphery. The fluorescein leakage associated with retinal edema may assume the petaloid configuration of cystoid macular edema or may be diffuse. Other fluorescein abnormalities include vascular loops and intraretinal shunts. The focus of treatment in patients with nonproliferative diabetic retinopathy and no macular edema is treatment of hyperglycemia and intercurrent systemic disease. A controlled clinical trial has shown that aldose reductase inhibitor therapy does not prevent progression of diabetic retinopathy. Focal argon laser treatment of discrete points of retinal leakage in patients with clinically significant macular edema, principally defined as thickening of the retina at or within 500 m of the center of the macula, reduces the risk of visual loss and increases the likelihood of visual improvement (see Chapter 24). Eyes with diabetic macular edema that is not clinically significant should usually be monitored closely without laser treatment. Since macular edema may be present with little or no change in visual acuity and requires slitlamp biomicroscopic retinal examination for full evaluation, primary health care providers should recognize the importance of prompt and early referral of diabetic patients to the ophthalmologist.
2. PROLIFERATIVE DIABETIC RETINOPATHY
The most severe ocular complications of diabetes mellitus are associated with proliferative diabetic retinopathy. Progressive retinal ischemia eventually stimulates the formation of delicate new vessels that leak serum proteins (and fluorescein) profusely. Neovascularization is frequently located on the surface of the disk and at the posterior edge of the peripheral zones of "nonperfusion" (Figures 10-24 and 10-25). Iris neovascularization, or rubeosis iridis, can also result.
The fragile new vessels proliferate onto the posterior face of the vitreous and become elevated once the vitreous starts to contract away from the retina. If the vessels bleed (Figure 10-26), massive vitreous hemorrhage may cause sudden visual loss. Eyes in which posterior vitreous detachment is complete are at less risk of developing neovascularization and vitreous hemorrhage. In eyes with proliferative diabetic retinopathy and persistent vitreoretinal adhesions, elevated neovascular fronds may undergo fibrous change and form tight fibrovascular bands that tug on the retina and exert continued vitreous contraction. This can cause either a progressive traction retinal detachment or, if a retinal tear is produced, rhegmatogenous retinal detachment. The retinal detachment may be heralded or concealed by vitreous hemorrhage. When vitreous contraction is complete in these eyes, proliferative retinopathy tends to enter the burned-out or "involutional" stage.
Treatment
Argon laser panretinal photocoagulation is usually indicated in proliferative diabetic retinopathy. Patients at greatest risk of significant visual loss are those with preretinal or vitreous hemorrhage or neovascularization of the disk. Panretinal photocoagulation can significantly reduce the chance of massive vitreous hemorrhage and retinal detachment in these patients by causing the regression and, in some cases, the disappearance of new vessels. The technique involves scattering up to several thousand regularly spaced laser burns throughout the retina, sparing the central region bordered by the disk and the major temporal vascular arcades (Chapter 24). Although the mechanism is not precisely understood, panretinal photocoagulation presumably works by reducing the angiogenic stimulus from ischemic retina.
The role of vitreoretinal surgery in proliferative diabetic eye disease continues to evolve. Conservative management of monocular vision impairing diabetic vitreous hemorrhage in the binocular patient had been to allow spontaneous resolution over the course of several months. The results of a 4-year study designed to assess the role of early vitrectomy for severe vitreous hemorrhage and proliferative diabetic retinopathy support this surgery as a means by which good vision may be restored or maintained. The role of vitreoretinal surgery in the treatment of diabetic traction retinal detachment is described elsewhere in this chapter.
CENTRAL RETINAL ARTERY OCCLUSION
The patient with central retinal artery occlusion routinely relates a history of painless catastrophic visual loss occurring over a period of seconds; antecedent transient visual loss (amaurosis fugax) may be reported. The visual acuity ranges between counting fingers and light perception in 90% of eyes at the time of initial examination. An afferent pupillary defect can appear within seconds after retinal arterial obstruction, preceding the fundus abnormalities by an hour.
Ophthalmoscopically, the superficial retina becomes opacified except in the foveola, where a cherry-red spot is evident (Figure 10-27). The cherry-red spot is pigment of the choroid and retinal pigment epithelium viewed through the extremely thin overlying foveolar retina and contrasted with the thicker and translucent perifoveolar retina. Twenty-five percent of eyes with central retinal artery occlusion have cilioretinal arteries that spare macular retina and may preserve some central visual acuity. Clinically, the retinal opacification resolves within 4-6 weeks, leaving a pale optic disk as the major ocular finding. In older patients, giant cell arteritis must be excluded and if necessary treated immediately with high doses of systemic corticosteroids. Other causes of central retinal artery occlusion are arteriosclerosis and emboli from carotid or cardiac sources. These are discussed further in Chapter 15.
Treatment
Because irreversible retinal damage has been shown to occur after 90 minutes of complete central retinal artery occlusion in the subhuman primate model, precious little time is available in which to begin therapy. Anterior chamber paracentesis can be employed in order to decrease intraocular pressure and increase retinal perfusion. This is particularly indicated in embolic central retinal artery occlusion. Intravenous acetazolamide has been used to decrease intraocular pressure, and an inhaled oxygen-carbon dioxide mixture has been employed to induce retinal vasodilation and increase the PO2 at the retinal surface. Direct infusion of a thrombolytic agent into the ophthalmic artery can result in recovery of vision. It must be performed within 8 hours after onset of the central retinal artery occlusion, requires specific radiologic expertise, and there is a risk of cerebral infarction. Systemic anticoagulants are generally not employed.
BRANCH RETINAL ARTERY OCCLUSION
Branch retinal artery occlusion usually presents with sudden loss of visual field and with reduction in visual acuity if the fovea is involved. Fundus signs of retinal edema with associated cotton-wool spots are limited to the area of retina supplied by the occluded vessel. Embolic causes are proportionately more common than in central retinal artery occlusion, and emboli are frequently identified on clinical examination (see Chapter 15). Migraine, oral contraceptive use, and vasculitis must also be considered.
CENTRAL RETINAL VEIN OCCLUSION
Central retinal vein occlusion is a common and easily diagnosed retinal vascular disorder with potentially blinding complications. The patient presents with sudden painless loss of vision. The clinical appearance varies from a few small scattered retinal hemorrhages and cotton-wool spots (Figure 10-28) to a marked hemorrhagic appearance with both deep and superficial retinal hemorrhage, which may rarely break through into the vitreous cavity. Most patients who develop the disease are over 50 years of age, and more than half have associated cardiovascular disease. Predisposing factors and their investigation are discussed in Chapter 15. Chronic open-angle glaucoma should always be excluded (see Chapter 11).
The two major complications associated with central retinal vein occlusion are reduced vision from macular edema and neovascular glaucoma secondary to iris neovascularization. Macular dysfunction occurs in almost all eyes with central vein occlusion. Although some eyes will show spontaneous improvement, most eyes will have persistent decreased central vision as a result of chronic macular edema. Nearly one-third of eyes with central retinal vein occlusion show significant retinal capillary nonperfusion on fluorescein angiography; one-half of these eyes will develop neovascular glaucoma.
Treatment
Careful follow-up evaluation is warranted, and prompt panretinal laser photocoagulation is recommended for eyes that develop anterior segment neovascularization. No treatment for macular edema, including grid pattern photocoagulation, has proved effective to date.
BRANCH RETINAL VEIN OCCLUSION
Branch retinal vein occlusion presents as sudden unilateral vision loss with segmentally distributed intraretinal hemorrhage. The vein occlusion always occurs at the site of an arteriovenous crossing (Figure 10-29), and retinal neovascularization may develop if the occlusion produces an area of retinal capillary nonperfusion that is more than 5 disk diameters in area. Sight-threatening complications of the disease are macular edema, macular ischemia, and vitreous hemorrhage from retinal neovascularization.
Treatment
Once peripheral retinal neovascularization has developed, sectoral laser retinal photocoagulation to the area of ischemic retina reduces the risk of vitreous hemorrhage by one-half. When vision loss due to macular edema persists for several months without spontaneous improvement, grid pattern argon laser macular photocoagulation may be indicated. Anticoagulant therapy has not been shown to be beneficial in either the prevention or the management of branch retinal vein occlusion. Investigation for an underlying systemic cause is discussed in Chapter 15. Important associated ocular diseases are chronic open-angle glaucoma and uveitis secondary to Behçet's syndrome.
RETINAL ARTERIAL MACROANEURYSM
Retinal macroaneurysms are fusiform or round dilations of the retinal arterioles occurring within the first three orders of arteriolar bifurcation. Most cases are unilateral, and the superotemporal artery is the most commonly involved vessel. Two-thirds of patients have associated systemic arterial hypertension.
The most common clinical symptom is loss of central vision as a result of retinal edema, exudation, or hemorrhage. Macroaneurysms may bleed into the subretinal space, into the retina, beneath the internal limiting membrane, or into the vitreous; the "hourglass" hemorrhage is typical and is due to bleeding beneath and anterior to the retina.
Although no clear indication for treatment with laser photocoagulation has been established, laser treatment of the macroaneurysm should be considered if lipid exudate coming from it threatens the fovea.
COLOR VISION DEFECTS
The perception of color is a cortical response to specific physical stimuli received by the retina. A narrow band of the electromagnetic spectrum, wavelengths between 400 and 700 nm, is capable of being absorbed by visual pigments contained in the outer segments of human cone photoreceptors. As described above, spectral sensitivity studies of cone photopigments have identified blue, green, and red cone photoreceptors. A minimal requirement for color discrimination is the presence of at least two kinds of cone photopigment, and normal color vision requires the presence of all three. Color vision testing is described in Chapter 2. In a broad sense, color vision defects are either congenital or acquired. While hereditary congenital color defects are almost always "red-green," affecting 8% of males and 0.5 % of females, acquired defects are more often of the "blue-yellow" variety and affect males and females equally. Congenital color vision defects affect both eyes equally, while acquired color defects frequently affect one eye more than the other. Most congenital color vision defects are X-linked recessive and are constant in type and severity throughout life. Acquired color vision defects generally vary in type and severity, depending upon the location and source of the usually ophthalmoscopically observable ocular pathology.
Dichromats are individuals whose cone photoreceptors contain only two of the three cone photopigments. Persons with a red-green color deficiency related to red-sensitive pigment loss were historically described first, and the condition is therefore referred to as protanopia. A second type of red-green deficiency involving green-sensitive pigment loss is known as deuteranopia. Blue-yellow color blindness is the third form and is referred to as tritanopia. While a color vision defect is present, there is no acuity loss in these patients.
Based on a color matching classification, the most common color vision deficit is that of anomalous trichromats. These individuals require three primaries for matching an unknown color but-unlike normal trichromats-use them in "anomalous" amounts. Each of the anomalous trichromats has a defect analogous to that of the dichromats described above.
There are two forms of monochromatism, and although both leave the affected individual completely without color discrimination, they are two quite separate entities. In rod monochromatism, the individual is born without functioning cones in the retina, and such a loss accounts for the associated symptoms of low visual acuity, absent color vision, photophobia, and nystagmus. The generalized loss of cones in this condition is shown unequivocally by the photopic electroretinogram. In cone monochromatism, affected individuals with this extremely rare condition have no hue discrimination but do have normal acuity and no photophobia or nystagmus. Cone monochromats do have cone photoreceptors, but all the cones contain the same visual pigment.
II. TUMORS OF THE RETINA
PRIMARY BENIGN INTRAOCULAR TUMORS
Retinal Angioma*
Retinal hemangiomas occur as isolated tumors or associated with cerebellar hemangioblastomas, pancreatic cysts and carcinomas, renal cysts and carcinomas, and pheochromocytomas in von Hippel-Lindau syndrome (Figure 10-30). The retinal tumors are pink or red, endophytic, and usually supplied by a large feeder vessel. Juxtapapillary tumors are usually exophytic. Vision is affected by bleeding or exudation from the tumor vessels. Photocoagulation, diathermy, and cryotherapy are used to treat the retinal lesions.
Astrocytic (Glial) Hamartomas
Astrocytic hamartomas are translucent to whitish retinal and optic nerve head tumors most frequently associated with tuberous sclerosis (Bourneville's disease) (Figure 10-31). They may also be associated with neurofibromatosis-1 and -2 or may occur as isolated findings. These tumors are congenital. They may grow slowly and, as they mature, become calcified, acquiring a mulberry configuration.
PRIMARY MALIGNANT TUMORS OF THE RETINA
Retinoblastoma (Figure 10-32)
Retinoblastoma is a rare but life-endangering tumor of childhood. Two-thirds of cases appear before the end of the third year; rare cases have been reported at almost every age. Bilateral disease occurs in about 30% of cases. This is generally a sign of heritable disease, but up to one-third of heritable cases have purely unilateral disease. An allele within chromosomal band 13q14 controls both the heritable and nonheritable forms of the tumor. The normal retinoblastoma gene, present in every individual, is a suppressor gene or anti-oncogene. Individuals with the heritable form of the disease have one altered allele in every cell of the body; when the other allele in a developing retinal cell is affected by a spontaneous mutation, the tumor develops. In the nonheritable form of the disease, both alleles of the normal retinoblastoma gene in a developing retinal cell are inactivated by spontaneous mutation. Survivors of the heritable form of the disease (those 5% of new cases who had an affected parent or those who have had a germinal mutation) have almost a 50% chance of producing an affected child.
Retinoblastomas may exhibit outward (exophytic) or inward (endophytic) growth-either or both. The latter then extend into the vitreous (Figure 10-33). Both types gradually fill the eye and extend through the optic nerve to the brain and, less commonly, along the emissary vessels and nerves in the sclera to the orbital tissues. Occasionally, they grow diffusely in the retina, discharging malignant cells into the vitreous or anterior chamber, thereby producing a pseudoinflammatory process and mimicking retinitis, vitritis, uveitis, or endophthalmitis. Microscopically, most retinoblastomas are composed of small, closely packed, round or polygonal cells with large, darkly staining nuclei and scanty cytoplasm. They sometimes form characteristic Flexner-Wintersteiner rosettes, which are indicative of photoreceptor differentiation. Degenerative changes are frequent, accompanied by necrosis and calcification. A few will spontaneously resolve.
Retinoblastoma usually remains unnoticed until it has advanced far enough to produce a white pupil (leukocoria), strabismus, or inflammation. All children with strabismus or intraocular inflammation should be evaluated for the presence of retinoblastoma. The tumor is usually seen in the early stages only when sought for, as in children having a hereditary background or in cases where the other eye has been affected.
Retrolental fibroplasia, persistence of the primary vitreous, retinal dysplasia, Coats' disease, and nematode endophthalmitis may simulate retinoblastoma.
In general, the earlier the discovery and treatment of the tumor, the better the chance to prevent spread through the optic nerve and orbital tissues.
Enucleation is the treatment of choice for large retinoblastomas. Eyes with smaller tumors can be effectively treated with plaque or external beam radiotherapy (Figure 10-34), cryotherapy, or photocoagulation. Chemotherapy is being used to reduce the size of large tumors prior to other types of therapy and occasionally as the sole form of therapy. It is also used to treat tumors that have extended into the brain, orbit, or distally and may be used after enucleation in patients at high risk for such widespread disease.
Second primary malignant tumors, especially osteosarcomas, develop in a large number (estimates range from 20% to 90%) of survivors of the heritable form of retinoblastomas after a period of many years. These patients need to be carefully evaluated for the remainder of their lives.
LYMPHOMA
Intraocular lymphomas rarely occur in association with systemic lymphomas but are not uncommon as primary tumors, most often involving the retina and vitreous. They were formerly called ocular reticulum cell sarcomas but are now considered to be large cell lymphomas. They often mimic retinitis, vitritis, or uveitis; therefore, it is important to consider this tumor in the differential diagnosis of unexplained intraocular inflammation in an older patient.
Central nervous system involvement is the usual cause of death. Radiation plus chemotherapy is the treatment of choice and prolongs survival.
*See also Retinocerebellar Angiomatosis in Chapter 14.
REFERENCESList of Figures
| Figure 10-1: Age-related macular degeneration with discrete (small arrow) and large confluent (large arrow) macular drusen. | |
| Figure 10-2: Central serous chorioretinopathy with sensory retinal detachment (arrows) extending into the fovea. | |
| Figure 10-3: Fluorescein angiogram of central serous chorioretinopathy shows active disease with both a retinal pigment epithelial detachment (small arrows) and a sensory retinal detachment (large arrows). Two foci of inactive disease (open arrows) are also present. | |
| Figure 10-4: Flower-petal pattern of fluorescein dye in a patient with cystoid macular edema after cataract surgery. | |
| Figure 10-5: Presumed ocular histoplasmosis syndrome with active disease (large arrows) and an inactive pigmented macular scar (small arrow). Peripapillary pigmentation (curved arrow) is also present. | |
| Figure 10-6: The early fluorescein angiogram shows an inactive hypofluorescent scar (small arrow) and the characteristic lacy hyperfluorescence of subretinal neovascularization (open arrows). | |
| Figure 10-7: Late fluorescein leakage from macular subretinal neovascularization in a patient with presumed ocular histoplasmosis syndrome. | |
| Figure 10-8: Typical macular lesion of acute multifocal posterior placoid pigment epitheliopathy. | |
| Figure 10-9: Multiple angioid streaks (arrows) extend from the optic nerve. (Courtesy of University of California, San Francisco.) | |
| Figure 10-10: Myopic macular degeneration with choroidal vessels (arrows) visible through atrophic retinal pigment epithelium. | |
| Figure 10-11: Macular hole (large arrows) with surrounding sensory retinal detachment (small arrows). | |
| Figure 10-12: Epiretinal macular membrane elevates retinal vessels (arrow) and produces retinal striae. | |
| Figure 10-13: Traumatic choroidal rupture resulting in pigmented scar. A choroidal vessel (arrow) is visible through the scar. | |
| Figure 10-14: X-linked juvenile retinoschisis with typical superficial retinal cysts in the fovea. | |
| Figure 10-15: Cone dystrophy with depigmentation and a bull's-eye pattern to the macula. | |
| Figure 10-16: Fundus flavimaculatus with multiple irregular fleck lesions (arrow) involving the macula. | |
| Figure 10-17: Vitelliform dystrophy with a well-demarcated cyst-like macular lesion. | |
| Figure 10-18: Retinopathy of prematurity with stretching of the macula and straightening of retinal vessels. | |
| Figure 10-19: Retinitis pigmentosa with arteriolar narrowing and peripheral retinal pigment clumping. | |
| Figure 10-20: Background diabetic retinopathy with abundant macular exudate (open arrow), micro-aneurysms (small arrow), and intraretinal hemorrhage (large arrow). | |
| Figure 10-21: Fluorescein angiogram in nonproliferative diabetic retinopathy shows microaneurysms (arrow) and perifoveal retinal vascular changes. | |
| Figure 10-22: Late phase fluorescein angiogram shows hyperfluorescence typical of noncystoid diabetic macular edema. | |
| Figure 10-23: Fluorescein angiogram shows hypofluorescence from capillary drop-out (arrows) typical of ischemic diabetic maculopathy. | |
| Figure 10-24: A frond of neovascular tissue (arrows) is seen along the superotemporal vascular arcade in this eye with proliferative diabetic retinopathy. | |
| Figure 10-25: Fluorescein angiogram of proliferative diabetic retinopathy shows leakage from the neovascular tissue. The pinpoint areas of hyperfluorescence are micro-aneurysms. | |
| Figure 10-26: Proliferative diabetic retinopathy with preretinal hemorrhage obscuring the inferior macula. Macular exudate, microaneurysms, and intraretinal hemorrhages are also present. | |
| Figure 10-27: Acute central retinal artery occlusion with opaque white retina and attenuated vessels. (Courtesy of University of California, San Francisco.) | |
| Figure 10-28: Central retinal vein occlusion with extensive superficial retinal hemorrhage obscuring macular and optic nerve detail. | |
| Figure 10-29: Branch retinal vein occlusion involves the superotemporal vein. The point of obstruction (arrow) is at an arteriovenous crossing. | |
| Figure 10-30: Angiomatosis retinae of Von Hippel-Lindau disease (drawing). (Courtesy of F Cordes.) | |
| Figure 10-31: Retinal astrocytic hamartoma. | |
| Figure 10-32: Retinoblastoma as viewed through the pupil. | |
| Figure 10-33: Endophytic retinoblastoma. | |
| Figure 10-34: Retinoblastoma after radiotherapy. |
List of Tables
| Table 10-1: Anatomic classification of macular dystrophies. | |
| Table 10-2: Stages of retinopathy of prematurity |
10.1036/1535-8860.ch10